



 , Feng-Jie Jianga, Yu Ouc, Sheng Fua, Yu-Feng Chaa, Shun Zhanga, Zong-Yuan Liua, Wen Zhoua,b, Ai-Min Wangaa,b, Yong-Lin Wanga,b
, Feng-Jie Jianga, Yu Ouc, Sheng Fua, Yu-Feng Chaa, Shun Zhanga, Zong-Yuan Liua, Wen Zhoua,b, Ai-Min Wangaa,b, Yong-Lin Wanga,b
b Provincial Key Laboratory of Pharmaceutics in Guizhou Province, Guiyang 550004, China;
c Guiyang Women and Children's Hospital and Health Institute, Guiyang 550001, China
Acyclic nucleoside phosphonates (ANPs) have been approved by the FDA for the treatment of viral infection [1]. Among current anti-HBV agents, adefovir dipivoxil has potent in vitro and in vivo activity against HBV. In particular, it has shown an impressive ability to suppress replication of HBV that is resistant to nucleoside analogues such as lamivudine, emtricitabine and famciclovir [2, 3, 4]. However, the dose-limiting nephrotoxicity of adefovir and its potential to release the toxic carrier segments have limited its clinical use [5]. In recent studies on the pathological mechanism for nephrotoxicity of adefovir dipivoxil, it has been confirmed that the adefovir phosphoric acid pair of anions is efficiently transported by the human renal organic anion transporter 1 (hOAT1), amembrane transport protein localized in themembranes of proximal convoluted tubular epithelial cells. This transporting process presumably mediates the accumulation of adefovir in renal proximal tubules, and results in nephrotoxicity [6, 7]. Moreover, pharmacological studies revealed that non-steroidal anti-inflammation drugs (NSAIDs) are potent inhibitors of hOAT1 and exhibit protective effects against hOAT1-mediated cytotoxicity of antiviral agents [8].Based upon the above research findings, in our previous study, we obtained a series of mono L-amino acid ester, mono non-steroid anti-inflammation drug carboxylic ester derivatives of adefovir with more potent antiviral activity and lower renal cell toxicity than adefovir dipivoxil [9].
It has been confirmed that alamifovir and abacavir possess potent antiviral activity by a cellular mechanism of converting to active metabolites through structural transformation of O-desmethylation and oxidative deamination, respectively. This property has been ascribed to 4-methoxy phenylthio or cyclopropylamino structural fragment substitution at the 6- position of the purine ring [10, 11, 12, 13]. The above results inspired us to design and synthesize a series of novel mono L-amino acid ester, mono non-steroid anti-inflammation drug carboxylic ester derivatives of acyclonucleoside phosphonates with substituted thiophenol or amino substitution at the 6-position of the purine ring via the sub-structure combination method by using the reported adefovir mixed phosphonates as lead compounds, and we preliminarily evaluated their anti-HBV activity.
In this letter, nine mono L-amino acid ester, mono non-steroid anti-inflammation drug carboxylic ester derivatives of purine ring substituted acyclonucleoside phosphonates, 9a-i were obtained for the first time, and their anti-HBV activity were evaluated in HepG2 2.2.15 cells. It is expected that the results of the reported study might be able to afford some valuable information on new acyclic nucleoside phosphonates prodrug design.
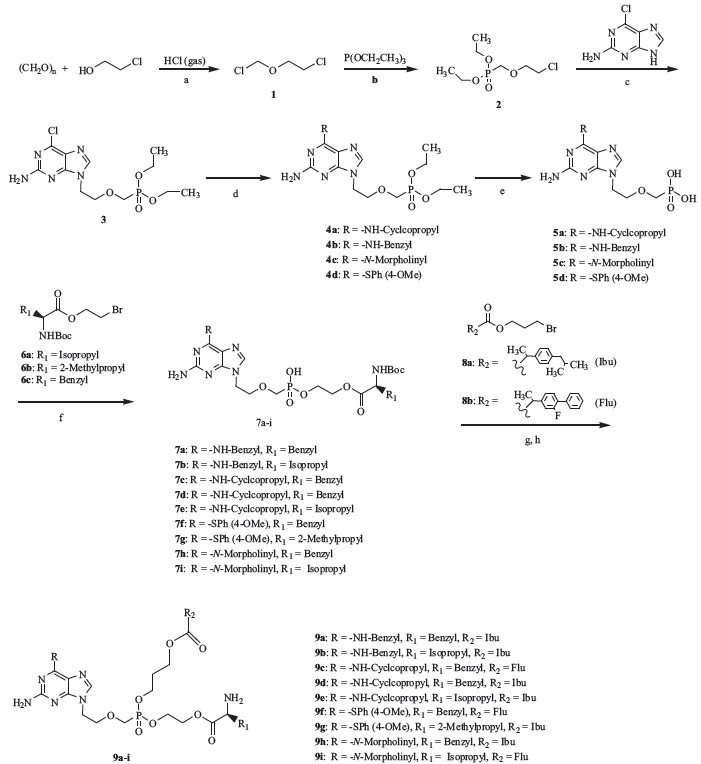
As shown in Scheme 1, paraformaldehyde was converted to 2- chloroethyl chloromethyl ether (1) by treatment with 2-chloroethanol and hydrochloric acid gas at low temperature (0 ℃-r.t.), the obtained compound then coupled with triethyl phosphite at 110 ℃ to afford 2-chloroethoxymethyl phosphonic acid diethyl ester (2). Then, condensation of the obtained 2 with 2-amino-6- chloropurine gave 2-amino-6-chloro-9-[2-(phosphonomethoxy) ethyl] purine bisethyl ester (3). The compound then coupled with substituted arylthiol, cyclopropylamine, benzylamine or morpholine in the presence of Et3N and DMF at 80-100 ℃ to afford compounds with different substitution at the 6-position of the purine ring (4a-d). These were then treated with trimethyl bromide silane at 80 ℃ to afford 2-amino-6-(arylthiol, cyclopropylamino, benzylamino or morpholinyl)-9-[2-(phosphonomethoxy) ethyl] purine (5a-d). The obtained compounds then coupled with N-Boc L-amino acid 2-bromoethyl esters (6a-c) to form 2-amino-6-substitution-9-[2-(phosphonomethoxy) ethyl] purine mono N-Boc L-amino acid ethyl ester (7a-i), which then coupled with 3-bromo-1-propyl esters of non-steroidal antiinflammatory drugs (8a,8b), in the presence of N,N'-dicyclo- $()1DFGIST[]cehm hexyL-4-morpholine carboxamidine (DCMC) as an acid scavenger in anhydrous DMF by using a "one pot synthesis" method. Finally, we removed the protecting group N-Boc by using 85% H3PO4 to obtain the target compounds (9a-i). Total yields for target compounds were in the range of 6.9%-15.2%.

|
Download:
|
| Scheme 1.Reagents and conditions: (a) 0 ℃–r.t., 12 h; (b) 110 ℃, 7 h; (c) anhydrous DMF, 80 ℃, 12 h; (d) substituted arylthiol, cyclopropylamine, benzylamine or morpholine, Et3N, anhydrous DMF, CsCO3, 80–100 ℃; (e) (CH3)3SiBr, 80 ℃, 12 h; (f) N,N-dicyclohexyl-4-morpholine carboxamidine (DCMC), anhydrous DMF, 60 ℃, 24 h; (g) DCMC, anhydrous DMF, 80–90 ℃, 48 h; and (h) 85% H3PO4/CHCl3, r.t., 6 h. | |
The synthesis: The preparation of the target compounds (9a-i) was similar to reported methods in Ref. [9]. 2-Amino- 6-substituted-9-[2-(phosphonomethoxy)ethyl]purine (5a-d) (0.47 mmol) and N,N'-dicyclohexyL-4-morpholine carboxamidine (DCMC) (0.94 mmol) were suspended in 40 mL anhydrous DMF, stirred at 45-55 ℃ until the mixture became homogeneous, and then a solution of 2-bromoethyl ester of L-amino acid (6a-c) (0.61 mmol) in 5 mL anhydrous DMF was added to the solution. The mixture stirred at that temperature for 24 h, until maximum conversion to 2-amino-6-substitution-9-[2-(phosphonomethoxy) ethyl]purine mono N-Boc L-amino acid ethyl ester (7a-i) which were not isolated from the mixture. Then, a solution of 3-bromo-1-propyl ester of ibuprofen or flurbiprofen (8a,8b) (0.94 mmol) in 5 mL anhydrous DMF was added to the above solution, respectively, and the reaction temperature was increased from 45-55 ℃ to 85-100 ℃. The reaction was continued at that temperature for 24-36 h. After that, the solvent was evaporated in vacuo. Ethyl acetate (50 mL) was added to the residue and the mixture was kept at 4-6 ℃ for 12 h, filtered, and concentrated in vacuo. The residue was purified by using routine flash column chromatography on silica gel (eluent:ethyl acetate/methanol = 15:1, v/v) to obtained mono N-Boc L-amino acid ester, mono NSAID carboxylic ester prodrugs of 6-substituted acyclonucleoside phosphonate. The deprotection process for the N-Boc group was similar to Ref. [9], by using aqueous phosphoric acid (85%) as a hydrolysis agent in chloroform medium at room temperature. When the reaction completed, the mixture was extracted by using chloroform, then the pH of the aqueous layer was adjusted to 7.0, and then it was extracted by using chloroform, The organic layers were combined, dried over anhydrous MgSO4, and filtered, and then the solvent was removed in vacuo to afford light yellow residues, which were purified by chromatography on silica-gel (eluent: ethyl acetate/methanol = 10:1-20:1, v/v) to obtained target compounds.
Spectra data of compounds 9a-9i: 9a: 1H NMR (400 MHz, CDCl3): δ 7.56 (s, 1H), 7.32-7.20 (m, 10H), 7.18 (d, 2H, J = 7.2 Hz), 7.09 (d, 2H, J = 8.0 Hz), 4.77 (brs, 2H), 4.28-3.99 (m, 11H), 3.84- 3.68 (m, 7H), 3.09-3.05 (m, 1H), 2.90-2.84 (m, 1H), 2.43 (d, 2H, J = 7.2 Hz), 1.92-1.79 (m, 3H), 1.47 (d, 3H, J = 7.2 Hz), 0.88 (d, 6H, J = 6.4 Hz). ESI-MS (m/z): 816.3 [M+H]+. HRMS calcd. for C42H55N7O8P [M+H]+: 816.3850, found: 816.3834. 9b: 1H NMR (400 MHz, CDCl3): δ 7.52 (s, 1H), 7.39-7.37 (m, 2H), 7.33-7.30 (m, 3H), 7.19 (d, 2H, J = 8.0 Hz), 7.09 (d, 2H, J = 8.0 Hz), 4.78 (brs, 2H), 4.28-4.11 (m, 11H), 4.04-3.99 (m, 2H), 3.86-3.83 (m, 2H), 3.76- 3.65 (m, 3H), 2.43 (d, 2H, J = 7.2 Hz), 2.05-2.01 (m, 1H), 1.96-1.92 (m, 2H), 1.50 (d, 3H, J = 7.2 Hz), 0.96 (d, 3H, J = 7.2 Hz), 0.90-0.87 (m, 9H). ESI-MS (m/z): 768.3 [M+H]+. HRMS calcd. for C38H55N7O8P [M+H]+: 768.3844, found: 768.3859. 9c: 1H NMR (400 MHz, CDCl3): δ 7.56 (s, 1H), 7.53-7.51 (m, 2H), 7.45-7.33 (m, 4H), 7.31-7.21 (m, 3H), 7.20-7.09 (m, 4H), 4.91 (brs, 2H), 4.29-4.01 (m, 10H), 3.83-3.70 (m, 5H), 3.10-3.07 (m, 1H), 2.90-2.85 (m, 1H), 1.94-1.89 (m, 2H), 1.48 (d, 3H, J = 7.2 Hz), 1.25 (m, 1H), 0.86 (m, 2H), 0.62-0.59 (m, 2H). ESI-MS (m/z): 804.3 [M+H]+. HRMS calcd. for C40H48FN7O8P [M+H]+: 804.3281, found: 804.3243. 9d: 1H NMR (400 MHz, CDCl3): δ 7.56 (s, 1H), 7.31-7.21 (m, 5H), 7.19 (d, 2H, J = 8.4 Hz), 7.09 (d, 2H, J = 8.0 Hz), 5.38 (brs, 2H), 4.29-4.01 (m, 10H), 3.89-3.70 (m, 5H), 3.14-3.02 (m, 1H), 3.00-2.96 (m, 1H), 2.43 (d, 2H, J = 7.2 Hz), 2.02-1.81 (m, 3H), 1.48 (d, 3H, J = 7.2 Hz), 0.94-0.92 (m, 2H), 0.89 (d, 6H, J = 6.8 Hz), 0.66-0.60 (m, 2H). ESIMS (m/z): 766.3 [M+H]+. HRMS calcd. for C38H53N7O8P [M+H]+: 766.3688, found: 766.3661. 9e: 1H NMR (400 MHz, CDCl3): δ 7.54 (s, 1H), 7.19 (d, 2H, J = 8.0Hz), 7.09 (d, 2H, J = 8.4 Hz), 4.87 (brs, 2H), 4.27-4.11 (m, 10H), 4.05-4.00 (m, 2H), 3.85-3.80 (m, 2H), 3.75-3.67 (m, 3H), 2.44 (d, 2H, J = 7.2 Hz), 1.91-1.79 (m, 3H), 1.48 (d, 3H, J = 7.2 Hz), 0.89 (d, 12H, J = 6.4 Hz), 0.86-0.83 (m, 2H), 0.64-0.60 (m, 2H). ESI-MS (m/z): 718.3 [M+H]+. HRMS calcd. for C34H53N7O8P [M+H]+ : 718.3688, found: 718.3659. 9f: 1H NMR (400 MHz, CDCl3): δ 7.73 (s, 1H), 7.53-7.49 (m, 4H), 7.44-7.34 (m, 4H), 7.30-7.09 (m, 7H), 6.94 (d, 2H, J = 8.0 Hz), 4.86 (brs, 2H), 4.29-4.01 (m, 10H), 4.01-3.71 (m, 9H), 3.11-3.06 (m, 1H), 2.91- 2.86 (m, 1H), 1.96-1.90 (m, 2H), 1.53 (d, 3H, J = 7.2 Hz). ESI-MS (m/z): 887.2 [M+H]+. HRMS calcd. for C44H49FN6O9PS [M+H]+: 887.2998, found: 887.2967. 9g: 1H NMR (400 MHz, CDCl3): δ 7.57 (s, 1H), 7.54 (d, 2H, J = 8.8 Hz), 7.19 (d, 2H, J = 8.0Hz), 7.08 (d, 2H, J = 7.6 Hz), 6.96 (d, 2H, J = 8.8 Hz), 4.80 (brs, 2H), 4.28-4.14 (m, 8H), 4.04-4.03 (m, 2H), 3.84-3.69 (m, 5H), 3.72-3.67 (m, 2H), 2.42 (d, 2H, J = 7.2 Hz), 1.95-1.92 (m, 2H), 1.85-1.75 (m, 3H), 1.55-1.52 (m, 4H), 0.94-0.87 (m, 12H). ESI-MS (m/z): 815.3 [M+H]+. HRMS calcd. for C39H56N6O9PS [M+H]+: 815.3562, found: 815.3541. 9h: 1H NMR (400 MHz, CDCl3): δ 7.54 (s, 1H), 7.30 (d, 2H, J = 8.0 Hz), 7.24-7.16 (m, 5H), 7.08 (d, 2H, J = 8.0 Hz), 4.71 (brs, 2H), 4.26-4.09 (m, 11H), 4.05-4.01 (m, 2H), 3.85-3.83 (m, 2H), 3.79-3.72 (m, 8H), 3.11-3.08 (m, 1H), 2.92-2.88 (m, 1H), 2.90 (d, 2H, J = 8.0 Hz), 1.91-1.88 (m, 2H), 1.84-1.81 (m, 1H), 1.47 (d, 3H, J = 7.2 Hz), 0.89 (d, 6H, J = 6.4 Hz). ESI-MS (m/z): 796.3 [M+H]+. HRMS calcd. for C39H55N7O9P [M+H]+: 796.3793, found: 796.3814. 9i: 1H NMR (400 MHz, CDCl3): δ 7.55-7.51 (m, 3H), 7.39-7.34 (m, 4H), 7.15-7.09 (m, 2H), 4.71 (brs, 2H), 4.27-4.15 (m, 11H), 4.08-4.03 (m, 2H), 3.86-3.82 (m, 2H), 3.79-3.72 (m, 8H), 2.04-2.02 (m, 1H), 1.97-1.92 (m, 2H), 1.55 (d, 3H, J = 7.2 Hz), 0.97 (d, 3H, J = 6.8 Hz), 0.91 (d, 3H, J = 6.8 Hz). ESI-MS (m/z): 786.3 [M+H]+. HRMS calcd. for C37H50FN7O9P [M+H]+: 786.3386, found: 786.3362.
The synthesized compounds were evaluated for their inhibitory effect on the replication of HBV in HepG2 2.2.15 cells according to Ref. [14]. The HepG2 2.2.15 cells were plated at a density of 5 × 104 cells/well on 24-well plates and incubated for 2 days. The medium was replaced with medium containing test compounds in gradient concentration (10, 1, 0.1 mg/mL). After 2 days of treatment, the culture medium was removed, and then fresh medium containing the same concentration of the test compounds was added to the cultures again. At day 10, the supernatants were collected and lyzed for the intracellular HBV DNA analysis. The amount of HBV DNA in each condition was measured by real time PCR using Icycler (Bio-Rad). Amplification primers were HBVFP: 5'- TGT CCT GGTTAT CGC TGG-3' and HBVRP: 5'-CAA ACG GGC AAC ATA CCT T-3' . The TaqMan probe was FAM-5'-TGT GTC TGC GGC GTT TTA TCAT-3'-TAMRA. The half maximum cytotoxic concentration values (CC50) for each compound were measured by calculating the cell growth rates by a standard procedure. The cells were plated at a density of 5 × 104 cells/well on 24-well plates and incubated for 12 h. Then supernatant was replaced with DEME medium containing test compounds at final concentrations of 1000, 100, and 10 μmol/L and the cells were cultured for 2 more days. At the end of the incubation period, 10 μL of Cell Counting Kit-8 (CCK-8) was added to each well and the cells were cultured for another 2 h. The OD value was measured at 450 nm. The data were analyzed by using Icycler IQ 3.0. IC50, CC50 and SI of these compounds are reported in Table 1. Adefovir dipivoxil was used as positive control.
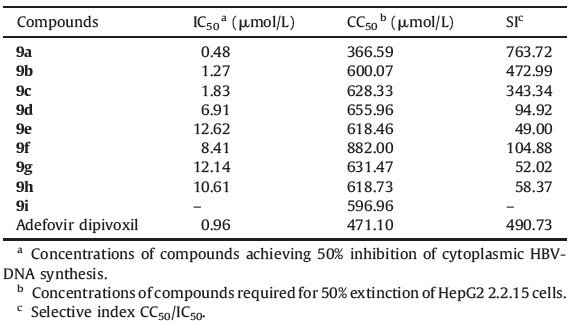
All synthesized compounds were screened for their inhibitory effects on the replication of HBV in HepG 2.2.15 cells. As indicated in Table 1, except for compound 9i, all synthesized compounds demonstrated anti-HBV activity with IC50 values of 0.48- 12.62 μmol/L. Compounds 9a, 9b, 9c, 9d and 9f exhibited potent antiviral activity with IC50 values range from 0.48 μmol/L to 8.41 μmol/L. Among them, compound 9a exhibited more potent antiviral activity than adefovir dipivoxil (IC50 0.96 μmol/L) with an IC50 value of 0.48 μmol/L, and the SI value of this compound (SI 763.72) was 1.5 times higher than that of the positive control (SI 490.72). The preliminary SAR research revealed that compounds with benzylamino substitution at the 6-position of the purine ring (9a, 9b) exhibited higher antiviral activity and selective index (SI) than those of compounds with cyclopropylamino substitution at the same position (9c-e ), and the latter have higher antiviral activity than that of compounds with 4-methoxy phenylthio (9f, 9g) or N-morpholinyl (9h,9i) substitution at the 6-position of the purine ring. Moreover, comparing the antiviral activity of target compounds with different L-amino acid fragments, it could be seen that compounds containing L-phenylalanine fragment (9a, 9c, 9d and 9f) possessed higher antiviral activity than compounds containing an L-valine or L-leucine fragment (9b, 9e and 9g).
| Table 1 Anti-HBV evaluation of mono L-amino acid ester, mono NSAIDs carboxylic ester prodrugs of 6-substituted acyclonucleoside phosphonates. |
{kind=link}
In summary, a new series of mono L-amino acid ester, mono non-steroid anti-inflammation drug (NSAID) carboxylic ester derivatives of acyclonucleoside phosphonates with substitution at the 6-position of purine were synthesized by using a "one pot synthesis" method. Their antiviral activity was evaluated in HepG 2.2.15 cells. Their renal cell toxicity and in vitro pharmacokinetical processes are still under investigation in our laboratory.
This work was supported by the National Natural Science Foundation of China (Nos. 20962004, 81260473), Projects of Guizhou Science and Technology Department (Nos. 2013-3031, 2012-3013), and Excellent Youth Scientific Talents Foundation of Guizhou (No. 2013-45).
| [1] | E. de Clercq, Clinical potential of the acyclic nucleoside phosphonates cidofovir, adefovir, and tenofovir in treatment of DNA virus and retrovirus infections, Clin. Microbiol. Rev. 16 (2003) 569-596. |
| [2] | A.K. Raney, R.K. Hamatake, Z. Hong, Agents in clinical development for the treatment of chronic hepatitis B, Expert Opin. Investig. Drugs 12 (2003) 1281-1295. |
| [3] | M.F. Yuen, C.L. Lai, Adefovir dipivoxil in chronic hepatitis B infection, Expert Opin. Pharmacother. 5 (2004) 2361-2367. |
| [4] | E. de Clercq, A. Holy, Acyclic nucleoside phosphonates: a key class of antiviral drugs, Nat. Rev. Drug Discov. 4 (2005) 928-940. |
| [5] | R. Valle, L. Haragsim, Nephrotoxicity as a complication of antiretroviral therapy, Adv. Chronic Kidney Dis. 13 (2006) 314-319. |
| [6] | E.S. Ho, D.C. Lin, D.B. Mendel, T. Cihlar, Cytotoxicity of antiviral nucleotides adefovir and cidofovir is induced by the expression of human renal organic anion transporter 1, J. Am. Soc. Nephrol. 11 (2000) 383-393. |
| [7] | D.H. Sweet, K.T. Bush, S.K. Nigam, The organic anion transporter family: from physiology to ontogeny and the clinic, Am. J. Physiol. Ren. Physiol. 281 (2001) F197-F205. |
| [8] | A.S. Mulato, E.S. Ho, T. Cihlar, Nonsteroidal anti-inflammatory drugs efficiently reduce the transport and cytotoxicity of adefovir mediated by the human renal organic anion transporter 1, J. Pharmacol. Exp. Ther. 295 (2000) 10-15. |
| [9] | X.Z. Fu, Y. Ou, J.Y. Pei, et al., Synthesis, anti-HBV activity and renal cell toxicity evaluation of mixed phosphonate prodrugs of adefovir, Eur. J. Med. Chem. 49 (2012) 211-218. |
| [10] | N. Kamiya, A. Kubota, Y. Iwase, et al., Antiviral activities of MCC-478, a novel and specific inhibitor of hepatitis B virus, Antimicrob. Agents Chemother. 46 (2002) 2872-2877. |
| [11] | S.K. Ono-Nita, N. Kato, Y. Shiratori, F.J. Carrilho, M. Omata, Novel nucleoside analogue MCC-478 (LY582563) is effective against wild-type or lamivudineresistant hepatitis B virus, Antimicrob. Agents Chemother. 46 (2002) 2602-2605. |
| [12] | E. Murakami, H. Bao, R.T. Mosley, et al., Adenosine deaminase-like protein 1 (ADAL1): characterization and substrate specificity in the hydrolysis of N(6)-or O(6)-substituted purine or 2-aminopurine nucleoside monophosphates, J. Med. Chem. 54 (2011) 5902-5914. |
| [13] | X.Z. Fu, Y. Ou, J. Xin, Y.S. Yang, Design, synthesis and in vitro evaluation of mono (2, 2, 2-trifluoro-ethyl) esters, mono L-amino acid ester prodrugs of acyclic nucleoside phosphonates as anti-HBV agents, Chin. Chem. Lett. 22 (2011) 1387-1390. |
| [14] | M.A. Sells, M.L. Chen, G. Acs, Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA, Proc. Natl. Acad. Sci. U.S.A. 84 (1987) 1005-1009. |