




During the past decades, infrared (IR) spectroscopy has been widely applied for studying secondary structures of peptides and proteins in the condensed phase [1]. Compared to the vibrational properties in the gas phase, the phenomena in the aqueous phase become more complicated due to the formation of intermolecular hydrogen bonds with surrounding water molecules. Theoretical interpretations of the IR spectroscopy can offer a deep understanding of the nature of the origins of various vibrational features in the aqueous phase at a molecular level [2, 3, 4]. However, traditional harmonic frequency calculations essentially suffer from some well-known problems, in which the vibrational anhamonicities, dynamics and temperature are not taken into account [4, 5].
Alternatively, molecular dynamics (MD) simulation with proper force fields can directly reproduce the experimental characteristics of IR spectroscopy of biomolecules in solution through the Fourier transform of dipole autocorrelation functions, since the vibrational anhamonicities, dynamics and temperature have been included in a direct way [4]. Nevertheless, effective atomic charges are always fixed in the common non-polarizable potentials, such as AMEBR [6, 7], OPLS-AA [8, 9], and CHARMM27 [10]. In fact, themagnitude of atomic charges presents a dynamical fluctuation responding to the changes of both the structure of the biomolecule itself, and the surrounding solvent environment. Consequently, such nonpolarizable potentials often fail to capture the dynamical electrostatic polarization (EP) of each atom [11] and considerable uncertainty of the calculated IR spectra may arise from the fact that the EP effects are not clear.
As one of simplest peptides, N-methylacetamide (NMA) is composed of a single amide group terminated by methyl group, making it a prototype model for characterizing the physical properties of the protein backbone [12, 13, 14, 15]. Recently, Gaigeot and co-workers [16] applied the Car-Parrinello MD (CPMD) method to correctly reproduce all amide features of NMA in the 1000- 2000 cm-1 region. Although complete ab initio MD calculations implicitly include the EP effects, the computational cost increases steeply with the system size, so that current ab initio MD simulations are limited to the small space and time scales. For the proper study of IR spectra, however, adequate sampling is accompanied by the need to perform large numbers of QM calculations. Alternatively, the mixed quantum and molecular mechanical (QM/MM) method offers a more efficient computational tool, where the solvent molecules are often treated by the molecular mechanical method and only the solute molecule is described by the quantum mechanical method. For example, Yang and Cho [17] investigated the IR spectroscopy of NMA in water by theQM/MMMDsimulations. The relative peak intensities of amide Ⅰ-Ⅲ modes and the bandwidth of amide I mode are consistent with experimental results.
Herein, the IR spectra in the range 1000-2000 cm-1 of NMA in water are calculated by using MD simulation with high-level QM/ MMcorrections, where the NMA molecule is treated by the B3LYP/ 6-311++G** and MP2/6-311++G** levels of theory, respectively. In this work, we mainly focus on the effect of different QM levels on the vibrational features of IR spectra. We also estimate the importance of the dynamical fluctuation process of the EP behavior on the IR spectra.
The MD simulation was carried out for one NMA molecule plus 1500 water molecules by using the Tinker 5.0 code [18] in the NPT ensemble at 298 K and 1 atm. The periodic boundary condition was applied to all directions of the cubic simulation cell. All equations of motion of Newton were integrated using the velocity Verlet algorithm with a time step of 1 fs, which can yield good conservation of both energy and linear momentum. The desired temperature was maintained using the Berendsen method with the coupling time of 0.4 ps. The CHARMM27 force field [10] was used for the NMA molecule along with the TIP3P water model. The cutoff distance was set to 10Å , which can always be less than half the length of the cell to keep the minimum image convention. The long-range electrostatic interactions were calculated by the particle mesh Ewald method. The simulation was first run at 1.0 ns for equilibration, and then the coordinates were saved every 5 fs for the next 1.0 ns (totally yielding 200,000 frames for further analysis).
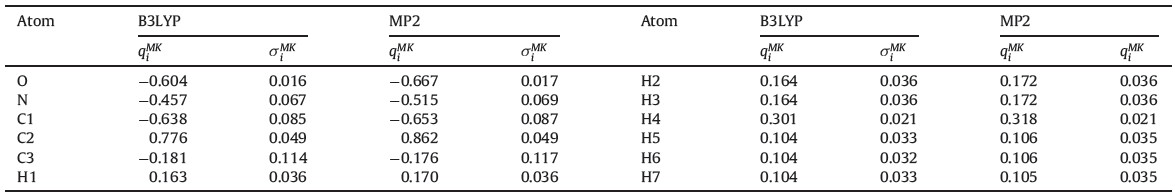
In order to incorporate the EP effects from the complicated solvent environment on the solute, the corresponding dipole moment surface of NMA was calculated from the combined QM/ MM method. The B3LYP/6-311++G** and MP2/6-311++G** levels of theory are used for the NMA molecule. As shown in Fig. 1a the radial distribution function of water molecules around the mass center of the NMA molecule shows an obvious multi-layer structure and the NMA/water interface region is up to r = 10.5Å (i.e., the position of g(r) = 1). In this work, the subsystem of the QM/ MM calculation includes the NMA molecule and all water molecules in the NMA/water interface region (see Fig. 1b). For each saved frame, a single-point energy calculation for the NMA molecule was performed with the electronic embedding scheme of the ONIOM method with the TIP3P water molecules considered as the background point charges. Hence, the electronic structures of NMA were polarized by the disposition of surrounding water molecules. And then the atomic charges qi of NMA along the MD trajectories were obtained at the B3LYP and MP2 levels of theory. Since only the solute was treated by the QM calculation, the total dipole momentMof NMA was directly obtained for each frame. All ab initio calculations for various subsystems are performed with the GAUSSIAN 03 program. [19] Finally, the IR absorption coefficient a(w) can be expressed as [20]
where β = 1/(kBT), V is the volume, c is the speed of light in vacuum,
n(w) is the refractive index, M is the total dipolemoment. In Eq. (1),
the dipole autocorrelation function

|
Download:
|
| Fig. 1.(a) Radial distribution function of water molecules around the mass center of NMA molecule, and (b) illustration for the separation of QM/MM subsystem from the whole system. Space fill model: NMA molecule; Wireframe model: water molecules (green). | |
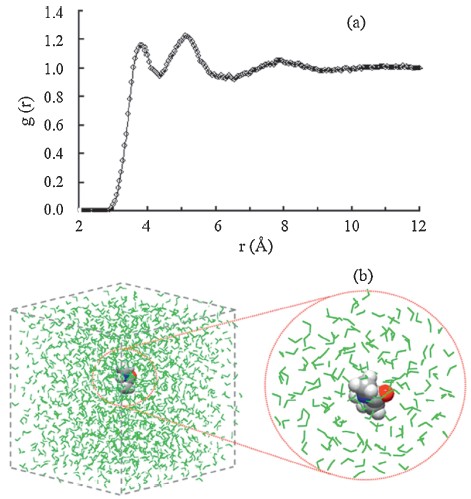
Wepresent the Merz-Kollman (MK) charge variations along the simulation time for several typical atoms of NMA molecule in Fig. 2. The MK atomic charges are obtained by fitting the electrostatic potentials from the self-consistent field density. According to the total dipole moment M = Σqir, the results from the MK atomic charges are well consistent with the NMA dipole moments. It should be noted that the total dipole moments obtained from the common Mulliken atomic charges are significantly different then the actual NMA results for most of saved frames. As shown in Fig. 2b and c the B3LYP and MP2 results display similar oscillations along the MD trajectories, suggesting the EP effect from surrounding solvent distribution is an obvious dynamical process rather than a constant value. Based on the B3LYP results, we can see from Table 1 that the standard deviations with respect to the average value are 0.016 e and 0.021 e for the O and H4 atoms, respectively. These deviations are much smaller than that of other atoms, such as 0.085 e for the C1 atom and 0.036 e for the H1 atom. Similarly, the smaller fluctuations of both the O and H4 charges along the simulation time can be compared to the C2 atom (see Fig. 2b and c). This difference can be attributed to the formation of hydrogen bonds (H-bonds) with surrounding water molecules. It is well known that there are two major types of intermolecular H-bonds in NMA-water complexes, i.e., C=O...H- O-H and N-H...OH2, respectively. The strong interactions from Hbonds lead to a relatively stable solvent environment around both the O and H4 atoms [22] so that the dynamical EP effects from surrounding water molecules are also relatively stable.

|
Download:
|
| Fig. 2.(a) Graphical representation of NMA molecule with the labels of each atom used in this work; the MK charge variations of several typical atoms from (b) the B3LYP results and (c) the MP2 results. | |
| Table 1 Ensemble averages qiMK and standard deviations qiMK of MK charge for each atom (labels as shown in Fig. 2a) of NMA molecule in the aqueous phase along classical MD trajectories from the QM/MM calculations, with the QM region treated by the B3LYP/6-311++G** and MP2/6-311++G** levels, respectively. |
{kind=link}
{kind=link}

|
Download:
|
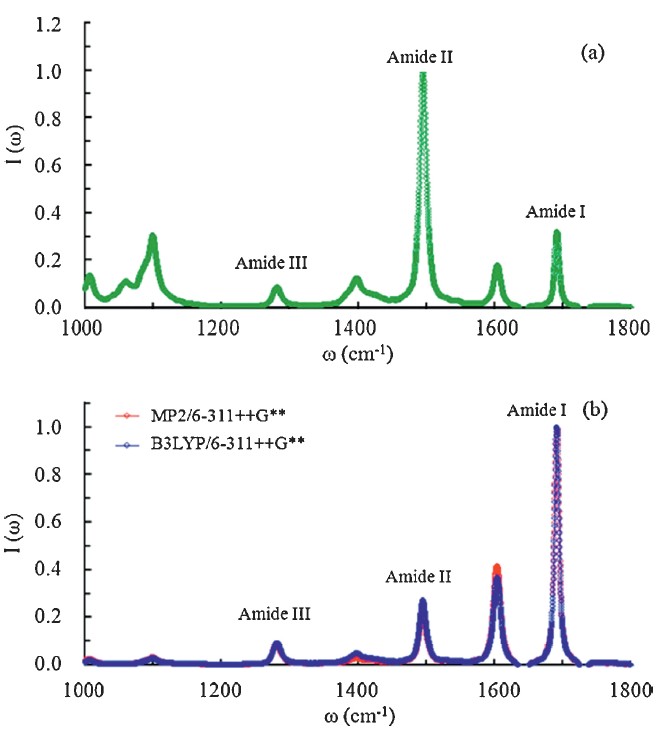
| Fig. 3.Normalized IR spectra ofNMAin water from classicalMDsimulation with the atomic charges from (a) the CHARMM27 model, (b) the QM/MM calculations. | |
{kind=link}
To summarize, the IR absorption spectra (1000-2000 cm-1) of the NMA molecule in water are calculated by combining the MD simulation with the high-level QM/MM corrections. To account for the EP effects, the B3LYP/6-311++G** and MP2/6-311++G** levels are used for the QM region to update the atomic charges of the NMA molecule for 200,000 saved MD frames. In this work, all B3LYP results are well consistent with the corresponding MP2 results, suggesting the B3LYP functional can give reasonable results for the IR spectra of peptides in the aqueous phase. By comparison, the calculated IR spectra from the CHARMM27 potential fail to simulate the correct relative intensities of amide Ⅰ-Ⅲ modes. On the contrary, the experimental relative intensities of amides Ⅰ-Ⅲ are well reproduced by the QM/MM corrected IR spectra. This work suggests that the consideration of the EP effects from solvent environments is necessary to reproduce reliable IR spectra of peptides and proteins in the aqueous phase.
This work was supported by the Natural Science Foundation of China (Nos. 21306070 and 20966003), and the Science & Technology Programs of Education Department of Jiangxi Province (No. GJJ12191). The authors are very grateful to Professor Shuhua Li (Nanjing University) for providing access to the supercomputers at Institute of Theoretical and Computational Chemistry, Nanjing University, China.
| [1] | H. Mantsch, D. Chapman, Infrared Spectroscopy of Biomolecules, Wiley-Liss, New York, 1996, pp. 1-3. |
| [2] | K.J. Jalkanen, V. Wurtz, A. Claussen, et al., The use of vibrational spectroscopies to study protein and DNA structure, hydration, and binding of biomolecules: a combined theoretical and experimental approach, Int. J. Quantum Chem. 106 (2006) 1160-1198. |
| [3] | J. Jeon, S. Yang, J.H. Choi, M. Cho, Computational vibrational spectroscopy of peptides and proteins in one and two dimensions, Acc. Chem. Res. 42 (2009) 1280-1289. |
| [4] | M.P. Gaigeot, Theoretical spectroscopy of floppy peptides at room temperature. A DFTMD perspective: gas and aqueous phase, Phys. Chem. Chem. Phys. 12 (2010) 3336-3359. |
| [5] | H. Jiang, R.R. Chen, H.L. Pu, et al., Studies on the binding of vinpocetine to human serum albumin by molecular spectroscopy and modeling, Chin. Chem. Lett. 23 (2012) 599-602. |
| [6] | Y. Duan, C. Wu, S. Chowdhury, et al., A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations, J. Comput. Chem. 24 (2003) 1999-2012. |
| [7] | V. Hornak, R. Abel, A. Okur, et al., Comparison of multiple amber force fields and development of improved protein backbone parameters, Proteins-Struct. Funct. Bioinform. 65 (2006) 712-725. |
| [8] | W.L. Jorgensen, D.S. Maxwell, J. Tirado-Rives, Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids, J. Am. Chem. Soc. 118 (1996) 11225-11236. |
| [9] | G. Kaminski, R.A. Friesner, et al., Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides, J. Phys. Chem. B 105 (2001) 6474-6487. |
| [10] | A.D. MacKerell, D. Bashford, R.L. Dunbrack, et al., All-atom empirical potential for molecular modeling and dynamics studies of proteins, J. Phys. Chem. B 102 (1998) 3586-3616. |
| [11] | H. Yu, C.L. Mazzanti, T.W. Whitfield, et al., A combined experimental and theoretical study of ion solvation in liquid N-methylacetamide, J. Am. Chem. Soc. 132 (2010) 10847-10856. |
| [12] | T.W. Whitfield, G.J. Martyna, S. Allison, et al., Liquid NMA: a surprisingly realistic model for hydrogen bonding motifs in proteins, Chem. Phys. Lett. 414 (2005) 210-214. |
| [13] | T.W. Whitfield, J. Crain, G.J. Martyna, Structural properties of liquid N-methylacetamide via ab initio, path integral, and classical molecular dynamics, J. Chem. Phys. 124 (2006) 094503-094517. |
| [14] | T.W.Whitfield, G.J.Martyna, S. Allison, et al., Structure and hydrogen bonding in neat N-methylacetamide: classical molecular dynamics and Raman spectroscopy studies of a liquid of peptidic fragments, J. Phys. Chem. B 110 (2006) 3624-3637. |
| [15] | E. Harder, V.M. Anisimov, T. Whitfield, et al., Understanding the dielectric properties of liquid amides from a polarizable force field, J. Phys. Chem. B 112 (2008) 3509-3521. |
| [16] | M.P. Gaigeot, R. Vuilleumier, M. Sprik, et al., Infrared spectroscopy of N-methylacetamide revisited by ab initio molecular dynamics simulations, J. Chem. Theory Comput. 1 (2005) 772-789. |
| [17] | S. Yang, M. Cho, IR spectra of N-methylacetamide in water predicted by combined quantum mechanical/molecular mechanical molecular dynamics simulations, J. Chem. Phys. 123 (2005) 134503-134507. |
| [18] | J.W. Ponder, TINKER 5.0: Software Tools for Molecular Design, Washington University School of Medicine, Saint Louis, MO, 2009. |
| [19] | M.J. Frisch, et al., Gaussian 03, Revision D.01, Gaussian, Inc., . Wallingford, CT, 2004 |
| [20] | D.A. McQuarrie, Statistical Mechanics, Harper Collins Publishers, New York, 1976, pp. 499-507. |
| [21] | R. Ramirez, T. Lo'pez-Ciudad, P.P. Kumar, et al., Quantum corrections to classical time-correlation functions: Hydrogen bonding and anharmonic floppy modes, J. Chem. Phys. 121 (2004) 3973-3983. |
| [22] | D. Laage, J.T. Hynes, Do more strongly hydrogen-bonded water molecules reorient more slowly? Chem. Phys. Lett. 433 (2006) 80-85. |
| [23] | X.G. Chen, R. Schweitzer-Stenner, S.A. Asher, N.G. Mirkin, S. Krimm, Vibrational assignments of trans-N-methylacetamide and some of its deuterated isotopomers from band decomposition of IR, visible, and resonance Raman spectra, J. Phys. Chem. 99 (1995) 3074-3083. |