




b Department of Chemistry, Dnyanopasak College, Parbhani 431401, MS, India;
c Department of Chemistry, Sharda Mahavidyalaya, Parbhani 431401, MS, India
One-pot multicomponant reactions are gaining more importance nowadays because of environmental implications [1]. MCRs are economically and environmentally advantageous because multi-step syntheses produce a considerable amount of waste mainly due to complex isolation procedures often involving expensive,toxic and hazardous solvents after each step [2]. Due to these advantages these reactions are perfectly suited for combinatorial library syntheses,and thus are finding increasing use in the discovery process for new drugs and agrochemicals [3].
Combinatorial chemistry is playing an increasingly important role as one of the tools inmodernmedicinal chemistry for the rapid discovery of new leads [4]. The preparation of libraries of small organic molecules is a rapidly evolving area of research [5]. Pyrimido pyrimidines are annulated uracils that have attracted considerable interest in recent years. Derivatives of pyrimido pyrimidine are known to display a wide range of pharmacological activities,such as antiviral [6],antibacterial [7],antitumor [8], anti-inflammatory [9],antifungal [10],and antileishmanial agent [11] and their potent binding affinity towards the tyrosine kinase domain of epidermal growth factor receptor [12] has also been demonstrated.
One-pot multicomponent reactions for the synthesis of pyrimido[4,5-d]pyrimidines are based on the condensation of carbonyl groups [13]. The Biginelli reaction [14] has been employed for the synthesis of pyrimido[4,5-d]pyrimidine by the one-pot condensation of acetophenone,urea and aldehydes and most of themethods involvesmodification of the Biginelli reaction by the condensation of aldehydes,urea and arylketones in acetic acid using a catalytic amount of KHSO4 [15]. Othermethods for the synthesis of pyrimido[4,5-d]pyrimidine using basic catalysts [16] or assisted by microwave irradiation[17] have been reported.
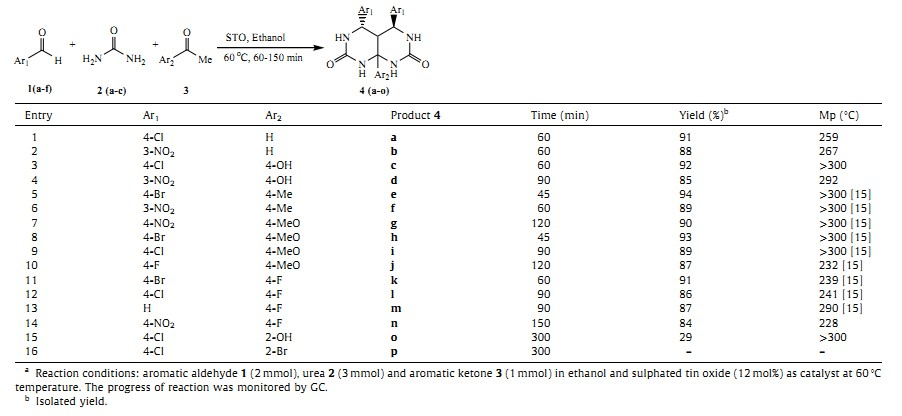
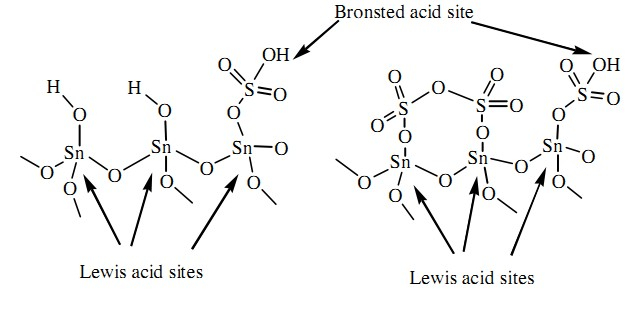
Recently sulfated metal oxides have been the focus of intensive research due to their high efficiency in organic transformations [18]. Sulfated metal oxide (SMO) is used as a catalyst in various organic syntheses [19]. Specifically,the sulfated tin oxide has been studied broadly. It has the strongest acidity on the surface [20]. It is a mesoporous material containing both Bronsted and Lewis acid sites (Fig. 1) [21]; it acts not only as a support,stabilizing the bonding molecular adsorbates,but also as a co-catalyst/activator, converting the neutral organometallic precursors into catalytically active electrophiles. It can be easily recovered from the reaction mixture by simple filtration and re-used after activation. In the earlier communications,our other research groups have demonstrated use of sulfated tin oxides in the synthesis of some valuable bioactive compounds [22].

|
Download:
|
| Figure. 1. Structure of sulfated tin oxide. | |
Owing to biologoical importance of pyrimido pyrimidines there has been remarkable interest for the preparation of these complex molecules in the synthetic organic chemistry. In continuation of our interest on synthesis of bioactive compounds [22] and solid heterogeneous catalysis [23],herein we describe an efficient and facile synthesis of uracil analogues with important biological activity. We examined the reaction of aromatic aldehydes, urea,and aromatic ketones using sulfated tin oxide as a solid heterogeneous catalyst to obtain pyrimido[4,5-d]pyrimidine derivatives.
2. ExperimentalAll solvents used were purchased from commercial venders without further purification. Aluminium sheets with a dimension of 20 cm × 20 cm,Silica gel 60 F254,Merck grade was used for thin layer chromatography tomonitor the progress of the reactions. The column chromatography was carried out over silica gel (80-120 mesh). Melting points were determined in open capillary tube and uncorrected. 1H NMR and 13C NMR spectra were recorded on an Avance 300 MHz spectrometer in DMSO-d6. Mass spectra were taken on Polaris-Q Thermoscintific GC-MS equipment. 2.1. General procedure for synthesis of sulfated tin oxide catalyst
A solution of 100 g stannous chloride (SnCl2 × nH2O) in 500 mL water was prepared with vigorous stirring. This solution was heated and a few drops of conc. HNO3 were added. After 1 h, heatingwas stopped and the reactionmixtureswas cooled to room temperature followed by the addition of a 25% ammonia solution with constant stirring until the pH value of the solution was adjusted to 8. The obtained precipitated product was collected by simple filtration and slowly poured into a 500 mL cold ammonium acetate solution (0.5-4%). The precipitated solid was filtered again and dried at 120 ℃ for 24 h. Tin oxide gel thus obtained was taken in a glass suction funnel and impregnated with 30 mL of concentrated sulfuric acid by slow addition; the resulting mixture was allowed to stand for 1 h. The solid catalyst was filtered and dried at 100 ℃ for 2 h. The catalyst was further heated in air at 500 ℃ for 3 h and stored in a sealed sample bottle until use.
4b: 1H NMR (300 MHz,DMSO-d6): δ 8.10-8.14 (m,2H,ArH), 7.98-8.06 (m,2H,ArH),7.77-7.90 (m,4H,ArH),7.32 (s,2H,NH), 7.35-7.41 (m,2H,ArH),7.22-7.29 (m,2H,ArH),6.86 (s,2H,NH), 5.18 (s,1H syn,CH),4.67 (d,1H anti, J = 6.9 Hz,CH),2.91-3.02 (m, 1H,CH); 13C NMR (75 MHz,DMSO-d6): δ 156.78,148.37,143.56, 139.69,129.87,128.81,125.43,124.89,123.45,56.48,54.71,45.11; GC-MS 488 m/z (M+); Anal. Calcd. for C24H20N6O6: C,59.01; H, 4.13; N,17.21; Found: C,59.04; H,4.15; N,17.24.
4c: H NMR (300 MHz,DMSO-d6): δ 8.27(s,1H,OH),7.54-7.63 (m,4H,ArH),7.38-7.49 (m,4H,ArH),7.25-7.31 (m,2H,ArH),7.11 (s,2H,NH),6.91-6.97 (m,2H,ArH),6.76 (s,2H,NH),5.24 (s,1H syn, CH),4.72 (d,1H anti,J = 6.8 Hz,CH),2.88-2.97 (m,1H,CH); 13C- NMR (75 MHz,DMSO-d6):δ 158.16,155.52,140.89,132.76, 130.27,129.48,127.37,116.71,55.88,53.86,42.46; GC-MS 483 m/z (M+); Anal. Calcd. for C24H20Cl2N4O3: C,59.64; H,4.17; N, 11.59; Found: C,59.67; H,4.21; N,11.61.
4d: 1H NMR (300 MHz,DMSO-d6): δ 8.31 (s,1H,OH),8.22-8.29 (m,2H,ArH),8.11-8.18 (m,2H,ArH),7.86-7.94 (m,2H,ArH),7.72- 7.80 (m,2H,ArH),7.41 (s,2H,NH),7.26-7.34 (m,2H,ArH),6.88- 6.95 (m,2H,ArH),6.79 (s,2H,NH),5.27 (s,1H syn,CH),4.82 (d,1H anti, J = 7.2 Hz,CH),3.08-3.11 (m,1H,CH); 13C NMR (75 MHz, DMSO-d6): δ 159.18,158.21,149.10,144.61,134.39,130.27, 125.93,123.64,116.03,57.59,55.63,43.75; GC-MS 504 m/z (M+); Anal. Calcd. for C24H20N6O7: C,57.14; H,4.00; N,16.66; Found: C, 57.12; H,3.98; N,16.68.
4n: 1H NMR (300 MHz,DMSO-d6): δ 8.46 (m,2H,ArH),8.32- 8.39 (m,2H,ArH),8.13-8.21 (m,2H,ArH),7.92-8.10 (m,2H,ArH), 7.82-7.90 (m,2H,ArH),7.54 (s,2H,NH),7.44-7.56 (m,2H,ArH), 6.67-6.72 (m,1H,ArH),6.88 (s,2H,NH),5.36 (s,1H syn,CH),4.89 (d,1H anti,J = 7.3 Hz,CH),2.91-3.01 (m,1H,CH); 13C NMR (75 MHz,DMSO-d6): δ 158.94,157.23,147.89,143.95,136.66, 129.49,126.73,124.37,115.29,57.62,55.47,44.19; GC-MS 506 m/z (M+); Anal. Calcd. for C24H19FN6O6: C,56.92; H,3.78; N,16.59; Found: C,56.94; H,3.81; N,16.62.
4o:1 H NMR (400 MHz,DMSO-d6): d 8.27(s,1H,OH),7.54-7.63 (m,4H,ArH),7.38-7.49 (m,4H,ArH),7.25-7.31 (m,2H,ArH), 7.11 (s,2H,NH),6.91-6.97 (m,2H,ArH),6.76 (s,2H,NH),5.24 (s,1H syn,CH),4.72(d,1H anti,J = 6.8 Hz,CH),2.88-2.97 (m,1H, CH); 13CNMR(75MHz,DMSO-d6): d 157.89,156.45,139.67, 130.34,130.08,128.42,127.88,115.94,56.17,54.04,41.73; GC- MS 483 m/z (M+); Anal. Calcd. for C24H20Cl2N4O3: C,59.64; H, 4.17; N,11.59; Found: C,59.65; H,4.15; N,11.58.
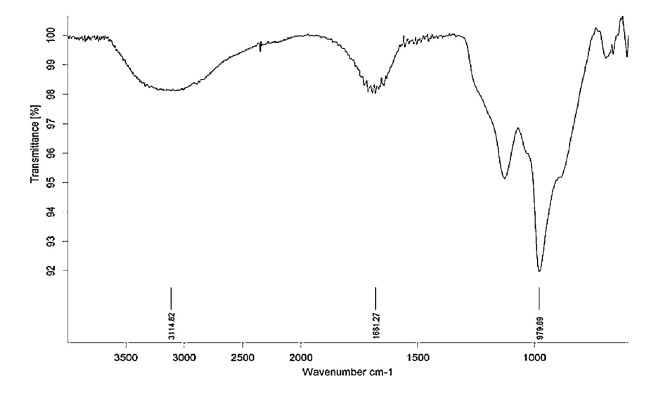
3. Results and discussionabThe sulfated tin oxide catalyst was prepared by well-established literature procedures [21]. The formation of the catalyst was confirmed by IR studies. The FT-IR spectra for the sulfated tin oxide shows the broad peaks in the region of 3400- 3000 cm-1 ,which can be attributed to the water adsorbed on the surface of the solid and the vibrational modes of OH bonds corresponding to the hydroxyl groups. Fig. 2 shows the vibrational mode of the sulfate groups at around 1200-900 cm-1 as a broad band,which confirmed the presence of the sulfate in the tin oxide. In the 600-400 regions the bands fromthe vibrationalmode of the Sn-O-Sn bonds were observed. Moreover,it is evident from literature that the coordination of sulfate groups and tin increases the specific surface area of the catalyst,stabilizing themesoporous structure and enhancing its thermal stability.

|
Download:
|
| Figure. 2.FT-IR spectrum of sulfated tin oxide. | |
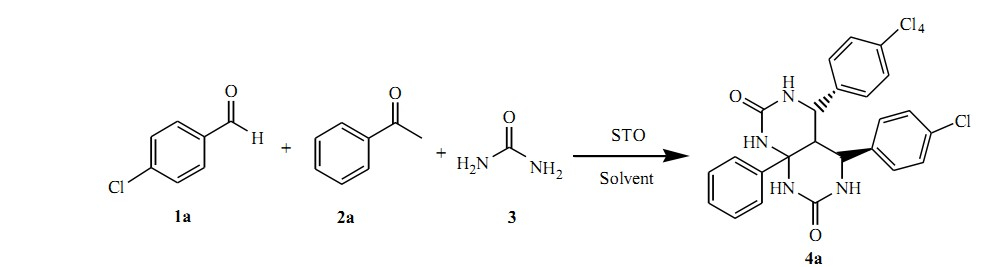
To evaluate the efficiency of the catalyst,the synthesis of 4,5,8a- triphenylhexahydropyrimido[4,5-d]pyrimidine-2,7(1H,3H)-dione was performed as a model reaction by the three-component condensation reaction of benzaldehyde,acetophenone and urea in the presence of varying catalytic amount of STO using various solvents (Scheme 1).

|
Download:
|
| Scheme 1.Optimization of synthesis of 4,5-di(4-chlorophenyl)-8a-phenylhexahydropyrimido[4,5-d]pyrimidine-2,7(1H,3H)-dione (<4a). | |
At first,to establish the optimal conditions,a set of experiments varying the amount of the catalysts and solvent at room temperature conditions were conducted. Initially when 5 mol% of sulfated tin oxide was used ethanol a low yield of the product 4a was obtained after 8 h of stirring (Table 1,entry 1). Increasing the catalyst quantity to 10 mol% improved the results. The reaction was completed in 5 h to produce 4a in good yield (67%) (Table 1, entry 2). Further increasing the quantity of the catalyst did not showany effect on reaction timewith an insignificant drop in yield (Table 1,entries 3 and 4).
| Table 1 Effect of catalyst quantity,solvent on the synthesis of 4a.a |

After optimization of catalytic quantity,further solvent screening was carried out to identify the best suitable solvent for the reaction. Under solvent free conditions,reaction to produce 4a took long time (6.5) to completewith a lowyield of 44% (Table 1, entry 5). Performing the reaction in aqueous media showed some improvement the over solvent free conditions with a better yield (62%) but small change in reaction time (6 h) was observed (Table 1,entry 6). When the reaction was carried out in polar organic solvent acetonitrile,although the reaction time was reduced,the yield of the product also dropped to 59% (Table 1, entry 7).Moreover organic non-polar solventswere deleterious for the reaction,requiring much longer time to complete in lowest yield (Table 1,entries 8 and 9). To check the necessity of the STO, the reaction was performed in the absence of the catalyst. Under such conditions very poor yield of 4a was obtained evenwithmuch extended reaction time (Table 1,entry 10).
In order to improve the performance of the reaction,the influence of temperature on the yield and reaction time was investigated. The results showed that,higher temperature was required for the synthesis of 4,5,8a-triphenylhexahydropyrimido- [4,5-d]pyrimidine-2,7(1H,3H)-dione 4a (Table 2). As mentioned before at room temperature after stirring for 300 min,the model reaction produced 4a in 67% yield,which was not reasonable (Table 2,entry 1). However,at slightly higher temperature of 40 ℃, the results showed a significant enhancement with a yield of 78% and a reduced reaction time of 240 min (Table 2,entry 2). Subsequently,when reaction was carried out at 50 ℃,reaction offered a superior yield (84%)with a boosted reaction rate (Table 2, entry 3). With a further incremental rise in temperature to 60 ℃, we achieved the desired optimum conditions. Reaction completed in relatively short period of time (60 min) to give an excellent yield (91%) of product 4a (Table 2,entry 4). Further elevation in temperature (70 8C) did show enrichment in terms of yield and reaction time as well (Table 2,entry 5).
| Table 2 Effect of temperature on the synthesis of 4a.a |

After optimization of the reaction conditions,the scope and generality of these conditions with other reactants were examined by using a variety of aromatic aldehydes,acetophenones and urea. The results are summarized in Table 3.
| Table 3 The synthesis of 4,5,8a-triarylhexahydropyrimido[4,5-d]pyrimidine-2,7(1H,3H)-dione 4.a |
Gratifyingly,we noted that both electron-withdrawing groups and electron-donating groups could tolerate the reaction conditions to give excellent yields (80%-96%). However,it was observed that the rate of reaction for aromatic compound containing electron-withdrawing groups was slower than compounds containing electron-donating groups in their aromatic rings. In the case of compound 4p no product was observed,which could have been due to the steric hindrance.
When using a solid acid catalyst,themost important point is the deactivation and recyclability of the catalyst. The catalyst was separated fromthe reactionmixture by a simple filtration followed by washing with an excess of distilled water and heating at 150 ℃ for 2 h. To study the recyclability of the catalyst,the STO catalyst was used for the same reaction repeatedly and the change in their catalytic activity was studied.
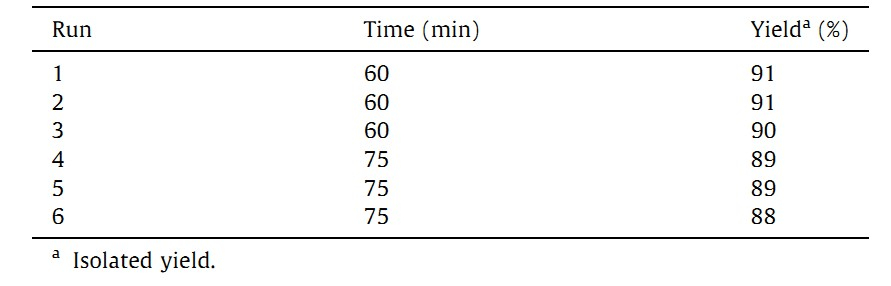
The relationship between the number of cycles of the reaction and the catalytic activity in terms of yields of product is presented in Table 4. The STO catalyst can be recycled and reused as a catalyst for at least five times with insignificant loss of activity. This slight decrease in the activity was due to partial leaching of the SO42, which leads to a decrease in the SO42 content in the solid catalyst.
| Table 4 Recyclability of catalyst sulfated tin oxide. |
{kind=link}
{kind=link}
{kind=link}
In conclusion,we have demonstrated the use of sulfated tin oxide as an efficient catalyst for the synthesis of 4,5,8a- triarylhexahydropyrimido[4,5-d]pyrimidine-2,7(1H,3H)-diones. The present method has several advantages compared to the previously reported protocols,for instant,high activity of catalysts, relatively low temperature,shorter reaction time,non hazardous reagents or solvent and higher chemical yield for a wide range of products. The known products are confirmed by their melting point,while for newly synthesized products both analytical and spectroscopic data are comparedwith similar compounds reported in the literature. Moreover,catalyst was reused for several runs with only an insignificant loss of activity.
AcknowledgmentsThe authors are thankful to the Principal Dr. P. L. More,DSM, College,Parbhani and Principal Dr. S. N. Thore,Deogiri College, Aurangabad,for encouragement during the process of carrying out this work.
| [1] | (a) C.C.A. Cariou, G.J. Clarkson, M. Shipman, Rapid synthesis of 1,3,4,4-tetrasubstituted b-lactams from methyleneaziridines using a four-component reaction, J. Org. Chem. 73 (2008) 9762-9764;(b) H.R. Shaterian, H. Yarahmadi, M. Ghashang, Silica supported perchloric acid (HClO4-SiO2): an efficient and recyclable heterogeneous catalyst for the one-pot synthesis of amidoalkyl naphthols, Tetrahedron 64 (2008) 1263-1269;(c) A. Znabet, E. Ruijter, F.J.J. de Kanter, et al., Highly stereoselective synthesis of substituted prolyl peptides using a combination of biocatalytic desymmetrization and multicomponent reactions, Angew. Chem. Int. Ed. 49 (2010) 5289-5292. |
| [2] | (a) A. Domling, I. Ugi, Multicomponent reactions with isocyanides, Angew. Chem. Int. Ed. 39 (2000) 3168-3210;(b) S. Heck, A.A. Domling, Versatile multi-component one-pot thiazole synthesis, Synlett 3 (2000) 424-426. |
| [3] | I. Ugi, A. Domling, Multicomponent reactions in organic chemistry, Endeavour 18 (1994) 115-122. |
| [4] | (a) E.M. Gordan, R.W. Barrett,W.J. Dower, S.P.A. Fodor,M.A. Gallop, Application of combinatorial technologies to drug discovery. 2. Combinatorial organic synthesis, library screening strategies and future directions, J. Med. Chem. 37 (1994) 1385-1401;(b) A.A. Virgilio, J.A. Ellman, Simultaneous solid-phase synthesis of β-turn mimetics incorporating side-chain functionality, J. Am. Chem. Soc. 116 (1994) 11580-11581;(c) S.M. Freier, D.A.M. Konings, J.R. Wyatt, D.J. Ecker, Deconvolution of combinatorial libraries for drug discovery: a model system, J. Med. Chem. 38 (1995) 344-352. |
| [5] | (a) E.K. Kick, J.A. Ellman, Expedient method for the solid-phase synthesis of aspartic acid protease inhibitors directed toward the generation of libraries, J. Med. Chem. 38 (1995) 1427-1430;(b) D.A. Campbell, J.C. Bermak, T.S. Burkoth, D.V. Patel, A transition state analog inhibitor combinatorial library, J. Am. Chem. Soc. 117 (1995) 5381-5382. |
| [6] | A.S. Jones, J.R. Sayers, R.T. Walker, E.D. Clercq, Synthesis and antiviral properties of (E)-5-(2-bromovinyl)-2'-deoxycytidine-related compounds, J. Med. Chem. 31 (1988) 268-271. |
| [7] | M.B. Deshmukh, S.M. Salunkhe, D.R. Patil, P.V. Anbhule, A novel and efficient one step synthesis of 2-amino-5-cyano-6-hydroxy-4-aryl pyrimidines and their antibacterial activity, Eur. J. Med. Chem. 44 (2009) 2651-2655. |
| [8] | C. Gasse, D. Douguet, V. Huteau, et al., Substituted benzyl-pyrimidines targeting thymidine monophosphate kinase of Mycobacterium tuberculosis: synthesis and in vitro anti-mycobacterial activity, Bioorg. Med. Chem. 16 (2008) 6075-6085. |
| [9] | R. Lin, S.G. Johnson, P.J. Connolly, et al., Synthesis and evaluation of 2,7-diaminothiazolo[ 4,5-d]pyrimidine analogues as anti-tumor epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors, Bioorg. Med. Chem. Lett. 19 (2009) 2333-2340. |
| [10] | E.P.S. Falcao, S.J. Melo, R.M. Srivastava, M.T.J.A. Catanho, S.C. Nascimento, Synthesis and antiinflammatory activity of 4-amino-2-aryl-5-cyano-6-{3- and 4-(Nphthalimidophenyl)} pyrimidines, Eur. J. Med. Chem. 41 (2006) 276-282. |
| [11] | Q. Chen, X.L. Zhu, L.L. Jiang, Z.M. Liu, G.F. Yang, Synthesis, antifungal activity and CoMFA analysis of novel 1,2,4-triazolo[1,5-a]pyrimidine derivatives, Eur. J. Med. Chem. 43 (2008) 595-603. |
| [12] | G.W. Rewcastle, A.J. Bridges, D.W. Fry, J.R. Rubin, W.A. Denny, Tyrosine kinase inhibitors. 12. Synthesis and structure-activity relationships for 6-substituted 4- (phenylamino)pyrimido[5,4-d]pyrimidines designed as inhibitors of the epidermal growth factor receptor, J. Med. Chem. 40 (1997) 1820-1826. |
| [13] | (a) D. Prajapati, P.P. Baruah, B.J. Gogoi1, J.S. Sandhu, One-pot synthesis of novel 1H-pyrimido[4,5-c] [1,2]diazepines and pyrazolo [3,4-d]pyrimidines Beilstein, J. Org. Chem. 2 (2006) 5, http://dx.doi.org/10.1186/1860-5397-2-5;(b) A. Bazgira, M.M. Khanaposhtani, A.A. Soorki, One-pot synthesis and antibacterial activities of pyrazolo[4',3':5,6]pyrido[2,3-d]pyrimidine-dione derivatives, Bioorg. Med. Chem. Lett. 18 (2008) 5800-5803. |
| [14] | P. Biginelli, Aldehyde-urea derivatives of aceto- and oxaloacetic acids, Gazz. Chim. Ital. 23 (1893) 360-413. |
| [15] | F. Shi, R. Jia, X. Zhang, et al., Extension of the Biginelli-type reaction: one-pot synthesis of pyrimidopyrimidines and spirobi[pyrimidine]s using potassium hydrogen sulfate as a catalyst, Synthesis 18 (2007) 2782-2790. |
| [16] | P. Sharma, N. Rane, V.K. Gurram, Synthesis and QSAR studies of pyrimido[4,5- d]pyrimidine-2,5-dione derivatives as potential antimicrobial agents, Bioorg. Med. Chem. Lett. 14 (2004) 4185-4190. |
| [17] | M. Dabiri, H. Arvin-Nezhad, H.R. Khavasi, A. Bazgir, Microwave-induced perchloric acid catalyzed novel solvent-free synthesis of 4-aryl-3,4-dihydropyrimidones via Biginelli condensation, J. Heterocycl. Chem. 44 (2007) 1009-1011. |
| [18] | (a) M. Hino, K. Atata, Synthesis of solid superacid catalyst with acid strength of H0≤ -16.04, J. Chem. Soc. Chem. Commun. 18 (1980) 851-852;(b) E. Igiesia, S.L. Sale, G.M. Kramer, Isomerization of alkanes on sulfated zirconia: promotion by Pt and by adamantyl hydride transfer species, J. Catal. 144 (1993) 238-253. |
| [19] | (a) B.M. Reddy, P.M. Sreekanth, P. Lakshmanan, Sulfated zirconia as an efficient catalyst for organic synthesis and transformation reactions, J. Mol. Cat. A: Chem. 237 (2005) 93-100;(b) B.M. Reddy, P.M. Sreekanth, An efficient synthesis of 1,5-benzodiazepine derivatives catalyzed by a solid superacid sulfated zirconia, Tetrahedron Lett. 44 (2003) 4447-4449;(c) X.B. Li, N. Katsutoshi, J.S. Laurent, O. Roberta, A.L. Johannes, Influence of calcination procedure on the catalytic property of sulfated zirconia, Catal. Lett. 113 (2007) 34-40. |
| [20] | J.C. Yori, M.A. Amato, G. Costa, J.M. Perera, Isomerization of n-butane on Pt/So42--ZrO2 and mechanical mixtures of Pt/Al2O3 + SO42--ZrO2, J. Catal. 153 (1995) 218-223. |
| [21] | (a) A.I. Ahmed, S.A. El-Hakam, A.S. KHder, W.S.A. El-Yazeed, Nanostructure sulfated tin oxide as an efficient catalyst for the preparation of 7-hydroxy-4- methyl coumarin by Pechmann condensation reaction, J. Mol. Cat. A: Chem. 366 (2013) 99-108;(b) T. Suzuki, T. Yokoi, R. Otomo, J.N. Kondo, T. Tatsumi, Dehydration of xylose over sulfated tin oxide catalyst: influences of the preparation conditions on the structural properties and catalytic performance, Appl. Catal. A: Gen. 408 (2011) 117-124. |
| [22] | (a) S.R. Sarda, V.B. Puri, A.B. Rode, et al., Sulfated tin oxides: a suitable reagent for synthesis of 2,4-diphenyl-4,6,7,8-tetrahydrochromen-5-one, Arkivoc 16 (2007) 246-251;(b) S.A. Dake, M.B. Khedkar, G.S. Irmale, et al., Sulfated tin oxide: a reusable and highly efficient heterogeneous catalyst for the synthesis of 2,4,5-triaryl-1Himidazole derivatives, Synth. Commun. 42 (2012) 1509-1520. |
| [23] | R.L. Magar, P.B. Thorat, V.B. Jadhav, et al., Silica gel supported polyamine: a versatile catalyst for one pot synthesis of 2-amino-4H- chromene derivatives, J. Mol. Cat. A: Chem. 374-375 (2013) 118-124. |