




b State Key Laboratory of Supramolecular Structure and Materials, Jilin University, Changchun 130024, China
1. Introduction
Organic semiconductors have received tremendous attention in recent years because of their potential application in organic light emitting diodes (OLED),organic solar cells and organic field-effect transistors (OFETs) [1, 2, 3]. Previous investigations demonstrate that designing molecules with donor-acceptor (D-A) moieties is a viable strategy to create organic semiconductors [4, 5, 6]. Zhang et al. report a new D-A-D molecule: BTBPD (Fig. 1) with bipyrrolylidene-2,2' (1H,1'H)-dione (BPD) as the electron-accepting core containing two benzo[b]-thiophene moieties [7]. The OFET of BTBPD exhibits an outstanding hole mobility of up to 1.4 cm2V-1s-1.

|
Download:
|
| Fig. 1. Molecular structure of BTBPD. | |
Recently,a great deal of effort has been invested in the theoretical study on typical photoelectric material systems to understand the experimental phenomenon and design new photoelectric materials [8, 9]. In order to gain insight into the origin of the hole transfer property of BTBPD,its charge transport features were systematically investigated by the Marcus theory in this article.
2. Computational methodsThe ground state and cationic state structures of BTBPD were fully optimized at density functional theory level with B3LYP functional and 6-31+g** basis set,which is recognized to successfully provide molecular geometries and accurately calculate the charge transport parameters for sulfur containing compounds [10, 11]. Harmonic vibrational frequencies were also calculated and confirmed that each optimized configuration was a minimum on the potential energy surface.
To describe the charge-transport properties of the compound, the Marcus electron transfer model was employed [12]. In this model,charge carrier diffuse by hopping from a charged molecule to an adjacent neutral one and each hopping step has been considered as a non-adiabatic electron-transfer reaction involving a self-exchange of charge between neighboring molecules. Thus the rate of charge transfer between neighboring molecules,kcan be expressed as:
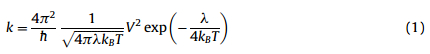
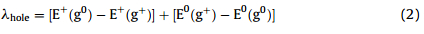
Thus,the drift mobility of hopping,μ,can be evaluated from the Einstein relation:

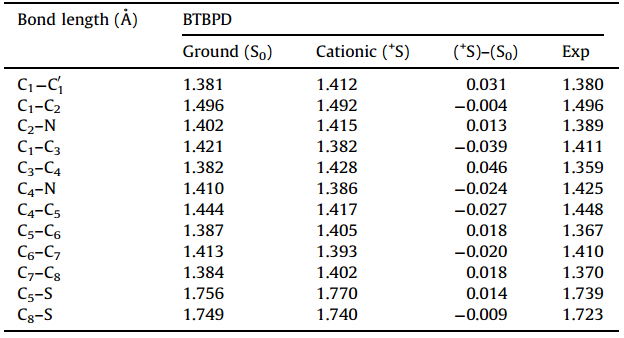
The schematic structure of BTBPD with the number of some key atom is shown in Fig. 1. The main geometrical parameters optimized at B3LYP level and the experimental results determined by the single-crystal X-ray diffraction are listed in Table 1.
| Table 1 Selected bond lengths ( Å ) in the ground and cationic state based on B3LYP |

|
Download:
|
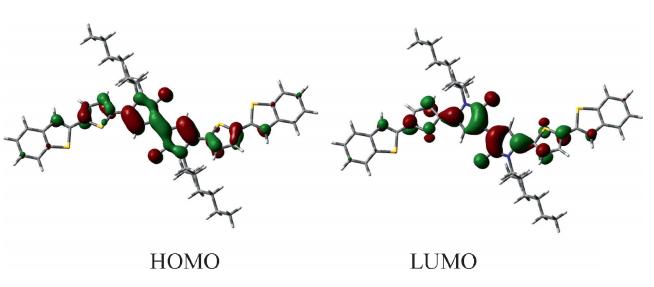
| Fig. 2. The frontier molecular orbitals calculated at B3LYP wave function. | |
In the cationic state,the molecular structure is almost coplanar. The thiophene rings form dihedral angles of 4.03°and 2.98° with BPD core and the benzo[b]thiophene rings,respectively. Moreover,the structural distortions are mainly localized on the central BPD core,such as,the bond length of C1-C3and C3-C4changes by-0.039 Å and 0.046 Å ,respectively. The degree of bond length alteration (BLA) in the central BPD core, calculated as the difference between the average bond length of C1-C3,C1-C2 and the average bond length of C1-C'1,C3-C4, amounts to 0.077 Å in the ground state and 0.017 Å in cationic state. The smaller BLA in the cationic state indicates that the p-electron are delocalized and creates an enhanced quinoidal character to the conjugated backbone in the cationic state, which facilitates the hole transport. The adiabatic potentialenergy surfaces of neutral/cation species were adopted to evaluate the reorganization energy. The calculated hole reorganization energy based on the DFT/B3LYP method is 0.496 eV, which is much close to that of the typical hole transport material TPD (0.28 eV) [13]. The small hole energy reveals that the structural distortions induced by hole injection are negligible, which is in agreement with the discussions above about the geometrical structure. Since the hole injection ability of hole transport materials is known to heavily depend on the ionization potential (IP),the calculated adiabatic IP (5.68 eV) and vertical IP (5.96 eV) indicate that the hole injection would be facile.
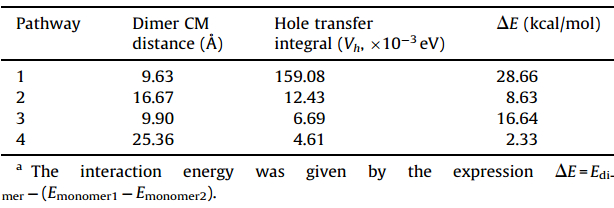
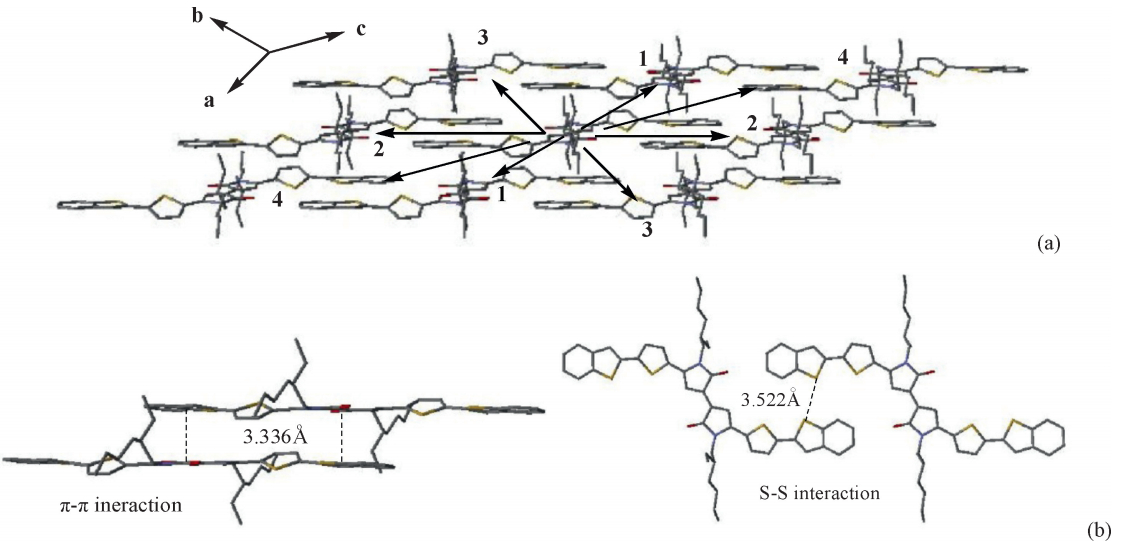
3.3. Transfer integralFig. 3 illustrates four main hopping pathways obtained directly from the crystal structure of BTBPD. The hole transfer integrals of (Vh) calculated with ADF at the PW91/TZP level of theory are listed in Table 2. The results indicate that the magnitudes of hole transfer integrals are sensitive to the molecular stacking arrangements in crystal. TheVhvalues in these pathways are large in the range of 10-3eV to 10-1eV. The statistical average value for Vh is 37.90 meV. In the parallel pathway 1 with theπ-πpacking orientation,theVh(159.08 meV) is the largest,indicating that the

|
Download:
|
| Fig. 3. (a) Main hopping pathways extracted from crystal and (b) π-π and S-S interaction existed in pathway 1 and 2 . | |
| Table 2 Calculated hole transfer integrals and corresponding intermolecular interactionDE for the major pathways.a |
{kind=link}
{kind=link}
{kind=link}
In addition,basis set superposition error (BSSE) using a counterpoise correction scheme was taken into account to calculate the interaction energies for main pathways. The results show high intermolecular interaction energy with the values of 28.66 kcal/mol and 8.63 kcal/mol occur on pathways 1and 2, respectively,in which theVhare big. This suggests that there is some correlation between the intermolecular interaction energy and the transfer integral.
3.4. Hole mobilityCombining the parameters mentioned above,we estimated the hole mobility by Marcus charge transfer model. The calculated hole mobility of BTBPD is large with a value of 0.29 cm2V-1s-1. ,which is ascribed to the smaller hole reorganization energy and large transfer integral. The calculated hole mobility is smaller than the experimental OFET result,which may be a result of the simplicity of the Marcus model. It is worth noting that this manuscript focuses on the structural and electronic properties in solid phase, and the accurate absolute hole mobility is not the aim of this work.So the Marcus theory could be still fully applicable,and the results presented in this work could shed light on materials design for organic semiconductors.
4. ConclusionIn summary,the structural,electronic and charge transport properties of BTBPD were systematically investigated by DFT calculations. The results show that the HOMO of BTBPD is mainly composed ofπ-orbital of the peripheral aromatic rings. The hole reorganization energy is small with a value of 0.496 eV. The hole transfer integrals of BTBPD are large,especially in the pathways 1 and 2 with π-π interaction and S···S interaction. Calculated hole drift mobility (0.29 cm2V-1s-1) suggests that BTBPD is a outstanding hole transport material and has potential applications in organic optoelectronic devices.
AcknowledgmentsThe authors acknowledge the financial support from the Ministry of Science and Technology of China (No. 09C26212203285) and the Project of Science and Technology of Jilin Province (No. 201115094).
| [1] | M. Bendikov, F. Wudl, D.F. Perepichka, Tetrathiafulvalenes, oligoacenenes, and their buckminsterfullerene derivatives: the brick and mortar of organic electronics, Chem. Rev. 104 (2004) 4891-4946. |
| [2] | J.E. Anthony, Functionalized acenes and heteroacenes for organic electronics, Chem. Rev. 106 (2006) 5028-5048. |
| [3] | V. Coropceanu, J. Cornil, D.A. da Silva Filho, et al., Charge transport in organic semiconductors, Chem. Rev. 107 (2007) 926-952. |
| [4] | S. Loser, C.J. Bruns, H. Miyauchi, et al., A naphthodithiophene-diketopyrrolopyrrole donor molecule for efficient solution-processed solar cells, J. Am. Chem. Soc. 133 (2011) 8142-8145. |
| [5] | O.P. Lee, A.T. Yiu, P.M. Beaujuge, et al., Efficient small molecule bulk heterojunction solar cells with high fill factors via pyrene-directed molecular self-assembly, Adv. Mater. 23 (2011) 5359-5363. |
| [6] | J.D. Yuen, J. Fan, J. Seifter, et al., High performance weak donor-acceptor polymers in thin film transistors: effect of the acceptor on electronic properties, ambipolar conductivity, mobility, and thermal stability, J, Am. Chem. Soc. 133 (2011) 20799-20807. |
| [7] | Z.X. Cai, Y.L. Guo, S.F. Yang, et al., New donor-acceptor-donor molecules with pechmann dye as the core moiety for solution-processed good-performance organic field-effect transistors, Chem. Mater. 25 (2013) 471-478. |
| [8] | X.D. Tang, Y. Liao, H.Z. Gao, Y. Geng, Z.M. Su, Theoretical study of the bridging effect on the charge carrier transport properties of cyclooctatetrathiophene and its derivatives, J. Mater. Chem. 22 (2012) 6907-6918. |
| [9] | Y. Geng, H.B. Li, S.X. Wu, Z.M. Su, The interplay of intermolecular interactions, packing motifs and electron transport properties in perylene diimide related materials: a theoretical perspective, J. Mater. Chem. 22 (2012) 20840-20851. |
| [10] | S.T. Bromley, M. Mas-Torrent, P. Hadley, C. Rovira, Importance of intermolecular interactions in assessing hopping mobilities in organic field effect transistors: pentacene versus dithiophene-tetrathiafulvalene, J. Am. Chem. Soc. 126 (2004) 6544-6545. |
| [11] | E.G. Kim, V. Coropceanu, N.E. Gruhn, et al., Charge transport parameters of the pentathienoacene crystal, J. Am. Chem. Soc. 129 (2007) 13072-13081. |
| [12] | R.A. Marcus, Electron transfer reactions in chemistry. Theory and experiment, Rev. Mod. Phys. 65 (1993) 599-610. |
| [13] | H.Z. Gao, Theoretical characterization of hole mobility in N,N'-diphenyl-N,N'-bis (3-methylphenyl)-(1,1'-biphenyl)-4,4'-diamine, J. Mol. Struct. Theochem. 962 (2010) 80-84. |