




b School of Chemistry and Chemical Engineering, Ningxia University, Yinchuan 750021, China
1. Introduction
Generally,the synthesis of tertiary amines from their parent trialkylamines is achieved by the two-step transformation involving dealkylation and re-alkylation. However,the selective removal of an alkyl group from the trialkylamine without preactivation is usually difficult. For example,demethylation from the tertiary amine is often incompatible with the requirements of high yield and mild reaction conditions[1] . The reaction of trialkylamines with alkyl halides can afford quaternary ammonium salts [2] ,and then the dealkylation of such salts might provide a way to the chemical modification of tertiary amines [3, 4, 5] . If the C-N bond cleaved in the subsequent de-quaternization is different from that formed in the quaternization,the transalkylation of tertiary amines with alkyl halides will be achieved. However,dequaternization has been reported mostly under high temperature [4] or using additional promoters [5] . Herein,we report a selective transalkylation of N-methyl tertiary amines with 3,4-dibromobutenolides under mild reaction conditions. 2. Experimental
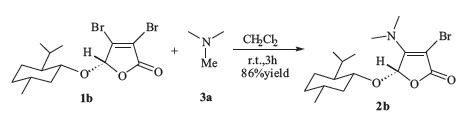
A typical procedure (Scheme 1) for the transalkylation by using trimethyl amine (3a) and 3,4-dibromobutenolide (1b) is presented as follows: To a stirred solution of 1b (396 mg,1.0 mmol) in CH2Cl2 (15 mL) was added 3a (303 mg,3.0 mmol). After 3 h at room temperature,the mixture was concentrated and the residue was purified by column chromatography to afford the desired enamine 2b as a colourless,dense oil (309 mg,86% yield): 1H NMR (400 MHz,CDCl3): δ 0.74 (d,3H,J = 6.8 Hz),0.80-0.86 (m,1H),0.88 (d,6H,J = 6.8 Hz),0.90-0.98 (m,1H),1.06-1.15 (m,1H),1.26-1.37 (m,2H),1.60-1.66 (m,2H),2.15-2.20 (m,2H),3.18 (brs,6H),3.51- 3.57 (m,1H),5.77 (s,1H); 13C NMR (100 MHz,CDCl3): δ 15.8,21.2, 22.2,22.8,25.0,31.7,34.0,40.8 (2),42.3,48.2,72.9,80.3,98.0, 159.3,168.8; IR (KBr,cm-1): ν 2957,1751,1646,1445,1113,746; HRMS (ESI): m/z Calcd. for C16H27BrNO3: 360.1169,found: 360.1166 [M+H]+. Experimental procedures,characterization data for all new compounds are available in Supporting information.

|
Download:
|
| Scheme. 1. General procedure for transalkylation of N-methyl tertiary amines. | |
Butenolides are very important units of many natural products and exhibit a broad range of biological activities[6] . The diverse synthesis of butenolides and their derivatives [7] for the evaluation of biological significance has attracted considerable attention from synthetic and medicinal chemists. Recently,we have introduced the thiadiazole moiety into the butenolides and some of gsubstituted butenolides showed good anticancer activities against HeLa cell lines [8, 9] . In order to get a new type of chiral butenolides for the examintion of anticancer activity,we decided to combine the butenolides with amino acids.
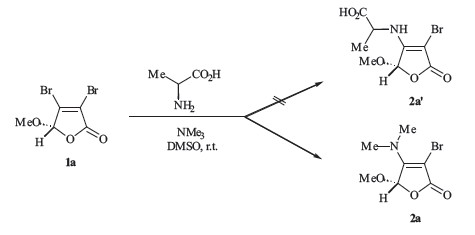
Initially,the known 3,4-dibromobutenolide 1a [8] and alanine were selected for this investigation (Scheme 2). In principle,the amino group of alanine could attack the electrophilic sites of butenolide 1a under basic conditions. The β-position of 1a would be more reactive towards the nucleophilic attack of amino group of alanine,in which a two-step sequence consisting of Michael addition and the following β-elimination of bromine would proceed readily. Surprisingly,the treatment of L-alanine with 3,4-dibromobutenolide 1a in the presence of trimethylamine in DMSO failed to give the expected butenolide 2a,. Interestingly, extensive spectra analysis (NMR,MS,HRMS and IR) suggested that the structure of the product was consistent with the enamine 2a. Further experiments performed in CH2Cl2 gave the enamine 2a in 87% yield. It is worthy to note that this transformation provided an alternative method for the transalkylation of tertiary amines,and the nitrogen unit from trimethylamine was effectively introduced into the butenolide skeleton.

|
Download:
|
| Scheme. 2. Proposed coupling of L-alanine with butenolide 1a. | |
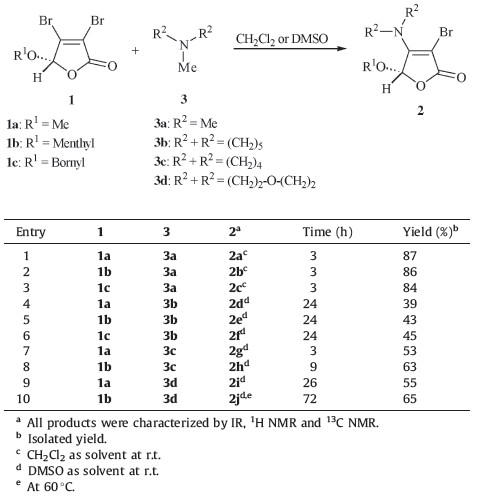
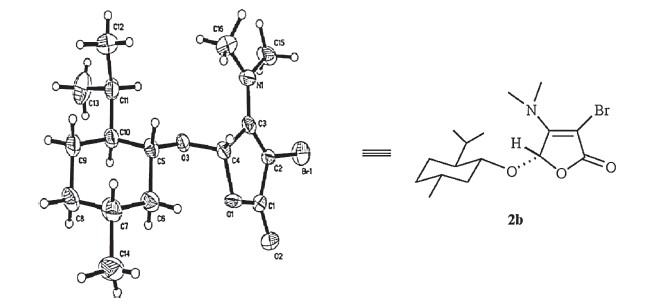
Encouraged by this result,two butenolides with chiral auxiliary, 1b and 1c, were then explored in the presence of trimethylamine for such transalkylation. As shown in Table 1,the reactions were proven to be successful,and the desired butenolides 2b and 2c (entries 2-3) were obtained in 86% and 84% yields,respectively. The enamine 2b could be recrystallized from the mixed solution of chloroform/ethanol (3:1),and the structure of the related product was then confirmed unambiguously by X-ray crystallographic analysis (Fig. 1). To expand the substrate scope,several different Nmethyl tertiary amines 3b-3d (entries 4-10) were subjected to current reaction,wherein using DMSO instead of CH2Cl2 promoted the reaction efficiency. Transalkylation of 1-methylpiperidine 3b with butenolides 1a-1c afforded the desired enamines 2d-2f in only moderate yields (entries 4-6). Compared with the results employing 1-methylpiperidine 3b (entries 4-6),the slightly improved conversions were observed in the cases using 1- methylpyrrolidine 3c (entries 7-8) and 4-methylmorpholine 3d (entries 9-10),and the corresponding heterocyclic butenolides 2g- 2j were obtained in 53-65% yields. According to the proposed mechanism,two competitive cleavages of C-N bond in the parent cyclic tertiary amines 3b-3d (entries 4-10) would exist,but the crude NMR analysis only showed the predominant formation of the desired enamines 2d-2j,which were obtained even in moderate yields. We thought it might be due to the low boiling point of methyl bromide,which was generated in situ and easy to release from the reaction mixture. Furthermore,the transalkylation of triethylamine or tripropylamine in the presence of 3,4- dibromobutenolides 1 were also preliminarily examined. Unfortunately, the desired enamine products 2 were always contaminated with some unknown impurities,demonstrating the substrate’s selectivity in the current transformation.
| Table 1 Amination of 3,4-dibromo-5-hydroxyfuran-2(5H)-one derivatives. |
{kind=link}
{kind=link}

|
Download:
|
| Fig. 1.X-ray structure of the enamine 2b. | |
{kind=link}
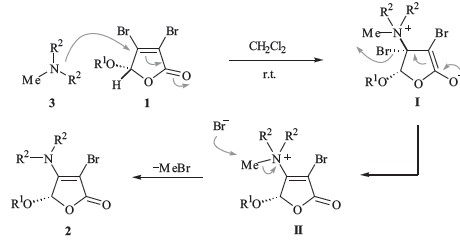
According to the above experimental results and literature reports,as depicted in Scheme 3,a one-pot process involving 1,4- conjuagte addition/β-elimination/dequaternization was proposed for this unusual selective transalkylation. First,1,4-conjugate addition of N-methyl tertiary amine 3 with Michael acceptor 1 would proceed to give the zwitterionic intermediate I,and then belimination of bromine would result in the formation of quaternary ammonium salt II. Subsequently,the selective cleavage of N-CH3 bond during the de-quaternization of the ammonium salt II would give the final enamine product 2 with a loss of volatile methyl bromide.

|
Download:
|
| Scheme. 3. Proposed mechanism for the selective transalkylation. | |
{kind=link}
The enamine product 2b (entry 2 of Table 1) has also been preliminarily tested for anticancer activity against HeLa cell lines. Compared with the butenolide analogues substituted with the thiadiazole and triazole moieties that we reported previously [8, 9] , the b-amino-substituted butenolide 2b showed a significantly improved anticancer effect with IC50 value of 0.19 μmol/L,giving a promising prospective for further biological investigation.
4. ConclusionIn conclusion,we have developed a one-pot protocol for the selective transalkylation of N-methyl tertiary amines with 3,4- dibromobutenolides,providing an alternative method for the synthesis of β-amino-substituted butenolides. The important feature of this novel method is that the given transalkylation could be achieved under very mild conditions,and no additional promoters were used. Currently,efforts to employ this transalkylation in the drug discovery are ongoing in our laboratory.
We thank NSFC (No. 21062014) and the ‘‘211’’ Project in Ningxia University for financial support.
| [1] | T. Rosenau, A. Hofinger, A. Potthast, P. Kosma, A general, selective, high-yield Ndemethylation procedure for tertiary amines by solid reagents in a convenient column chromatography-like setup, Org. Lett. 6(2004) 541-544. |
| [2] | Z. Zheng, T. Wu, R. Zheng, Y. Wu, X. Zhou, Study on the synthesis of quaternary ammonium salts using imidazolium ionic liquid as catalyst, Catal. Commun. 8(2007) 39-42. |
| [3] | (a) J. Cai, B. Wathey, A novel traceless solid phase tertiary amine synthesis based on Merrifield resin, Tetrahedron Lett. 42(2001) 1383-1385;(b) X.H. Ouyang, R.W. Armstrong, M.M. Murphy, A novel cleavage technique to generate small molecule compounds and libraries via a two-resin system, J. Org. Chem. 63(1998) 1027-1032;(c) A.R. Brown, D.C. Rees, Z. Rankovic, J.R. Morphy, Synthesis of tertiary amines using a polystyrene(REM) resin, J. Am. Chem. Soc. 119(1997) 3288-3295;(d) F.E.K. Kroll, J.R. Morphy, D.C. Rees, D. Gani, Resin-immobilised benzyl and aryl vinyl sulfones: new versatile traceless linkers for solid-phase organic synthesis, Tetrahedron Lett. 38(1997) 8573-8576;(e) P. Heinonen, H. Lonnberg, A novel solid support for derivatization and subsequent N-alkylation of secondary amines: preparation of N-alkylated 5-and 6-alkoxy-1,2,3,4-tetrahydroisoquinolines via mitsunobu reaction, Tetrahedron Lett. 38(1997) 8569-8572;(f) J.R. Morphy, Z. Rankovic, D.C. Rees, A novel linker strategy for solid-phase synthesis, Tetrahedron Lett. 37(1996) 3209-3212. |
| [4] | H.O. House, H.C. Miiller, C.G. Pitt, P.P. Wickham, Reduction of azabicyclic ketones, J. Org. Chem. 28(1963) 2407-2416. |
| [5] | (a) D.H.R. Barton, A. Fekih, X. Lusinchi, Sodium hydrogen telluride as a useful nucleophilic reagent for the cleavage of epoxides and of quaternary ammonium salts, Tetrahedron Lett. 26(1985) 6197-6200;(b) M.P. Cooke Jr., R.M. Parlman, Reduction of quarternary ammonium salts with lithium triethylborohydride. Convenient method for the demethylation of substituted trimethylammonium salts, J. Org. Chem. 40(1975) 531-532;(c) T.L. Ho, Dequaternization of ammonium salts by nucleophiles, Synth. Commun. 3(1973) 99-100;(d) R.O. Hutchins, F.J. Dux, Selective demethylation of quaternary salts with lithium propylmercaptide in hexamethylphosphoramide, J. Org. Chem. 38(1973) 1961-1962;(e) T.L. Ho, Dealkylation of quaternary ammonium salts with 1,4-diazabicyclo[2. 2. 2]octane, Synthesis 12(1972) 702;(f) T. Kametani, K. Kikasawa, M. Hiiragi, N. Wagatsuma, K. Wakisaka, Novel debenzylation of quaternary ammonium salts with thiophenol, Tetrahedron Lett. 10(1969) 635-638;(g) V. Simanek, A. Klasek, The demethylation of quarternary ammonium salts of some alkaloids by sodium selenophenolate, Tetrahedron Lett. 10(1969) 3039-3040. |
| [6] | (a) G.R. Flematti, E.L. Ghisalberti, K.W. Dixon, R.D. Trengove, Identification of alkyl substituted 2H-furo[2,3-c]pyran-2-ones as germination stimulants present in smoke, J. Agric. Food Chem. 57(2009) 9475-9480;(b) D. Tasdemir, G.P. Concepción, G.C. Mangalindan, et al., New terpenoids from a cacospongia sp. from the Philippines, Tetrahedron 56(2000) 9025-9030;(c) M.J. Ortega, E. Zubía, J.M. Ocaña, S. Naranjo, J. Salvá, New rubrolides from the Ascidian Synoicum blochmanni, Tetrahedron 56(2000) 3963-3967;(d) G.M. König, A.D. Wright, S.G. Franzblau, Assessment of antimycobacterial activity of a series of mainly marine derived natural products, Planta Med. 66(2000) 337-342;(e) K. Yoneyama, Y. Takeuchi, M. Ogasawara, et al., Cotylenins and fusicoccins stimulate seed germination of Striga hermonthica(Del.) benth and orobanche minor smith, J. Agric. Food Chem. 46(1998) 1583-1586;(f) Z. Jiang, D.Q. Yu, New type of mono-tetrahydrofuran ring acetogenins from Goniothalamus donnaiensis, J. Nat. Prod. 60(1997) 122-125;(g) S.L. Midland, N.T. Keen, J.J. Sims, Secosyrins 1 and 2 and syributins 1 and 2: novel structures produced by bacteria expressing the avrd gene, J. Org. Chem. 60(1995) 1118-1119;(h) J.N. Gnabre, J.L. Pinnas, D.G. Martin, et al., Characterization of steroidal glycosides from tylophora sylvatica, Tetrahedron 47(1991) 3545-3554;(i) S. Miao, R.J. Andersen, A.-H. Rubrolides, Metabolites of the colonial tunicate Ritterella rubra, J. Org. Chem. 56(1991) 6275-6280;(j) L.J. Reynolds, B.P. Morgan, G.A. Hite, E.D. Mihelich, E.A. Dennis, Phospholipase A2 inhibition and modification by manoalogue, J. Am. Chem. Soc. 110(1988) 5172-5177;(k) K. Kakinuma, J. Koike, K. Ishibashi, W. Takahashi, H. Takei, Structure-activity relationship and design of an antimutagen against the UV-Induced mutation of escherichia coli, Agric. Biol. Chem. 50(1986) 625-631;(l) M. Uchida, Y. Koike, G. Kusano, et al., Studies on the constituents of Chloranthus spp. Ⅲ. Six sesquiterpenes from Chloranthus japonicus, Chem. Pharm. Bull. 28(1980) 92-102. |
| [7] | (a) Y. Yaguchi, A. Nakahashi, N. Miura, et al., Stereochemical study of chiral tautomeric flavorous furanones by vibrational circular dichroism, Org. Lett. 10(2008) 4883-4885;(b) S.M. Ma, B. Wu, Z.J. Shi, An efficient synthesis of 4-halo-5-hydroxyfuran-2(5H)-ones via the sequential halolactonization and γ-hydroxylation of 4-aryl-2,3-alkadienoic acids, J. Org. Chem. 69(2004) 1429-1431;(c) S.P. Brown, N.C. Goodwin, D.W.C. MacMillan, The first enantioselective organocatalyticMukaiyama Michael reaction: a direct method for the synthesis of enantioenriched γ-butenolide architecture, J. Am. Chem. Soc. 125(2003) 1192-1194;(d) J. Jauch, A short total synthesis of kuehneromycin A, Angew. Chem. Int. Ed. 39(2000) 2764-2765;(e) T.E. Kedar, M.W. Miller, L.S. Hegedus, Synthesis of 4-alkyl-4-alkoxybutenolides having unsaturated side chains via chromiumcarbene complex photochemistry:(+)-cerulenin, J. Org. Chem. 61(1996) 6121-6126. |
| [8] | M.X. Wei, L. Feng, X.Q. Li, X.Z. Zhou, Z.H. Shao, Synthesis of new chiral 2,5-disubstituted 1,3,4-thiadiazoles possessing γ-butenolide moiety and preliminary evaluation of in vitro anticancer activity, Eur. J. Med. Chem. 44(2009) 3340-3344. |
| [9] | X. Li, X.Q. Li, H.M. Liu, X.Z. Zhou, Z.H. Shao, Synthesis and evaluation of antitumor activities of novel chiral 1,2,4-triazole Schiff bases bearing γ-butenolide moiety, Org. Med. Chem. Lett. 2(2012) 26. |