




b Depatment of Applied Chemistry, S.V. National Institute of Technology, Surat 395007, India;
c Doctoral Degree Program in Marine Biotechnology, National Sun Yat-Sen University, Kaohsiung 80424, Taiwan;
d Institute of Chemistry, Academia Sinica, Taipei 115, Taiwan;
e Institute of Cellular and Organismic Biology, Academia Sinica, Taipei 115, Taiwan;
f Center of Nanoscience and Nanotechnology, National Sun Yat-Sen University, Kaohsiung 80424, Taiwan;
g School of Pharmacy, College of Pharmacy, Kaohsiung Medical University, Kaohsiung 806, Taiwan
Acetophenone (AP) and its monohydroxy isomers (2-HAP,3- HAP and 4-HAP) are widely used as starting materials or intermediate chemicals for the synthesis of perfumes,drugs, and organo-metallic compounds. In addition,AP and 4-HAP exhibit some medicinal properties and are used as hypnotic and antiinflammatory activity agents [1] . AP analogues are also used as effective inhibitors of human cytosolic and mitochondrial aldehyde dehydrogenase. Regarding AP metabolism,the hydroxy position plays a key role in various biochemical pathways [2] and is also an important cholagogic constituent in bin hao (Artemisia scoparia) and as a cholagogue [3] . Moreover,4-HAP was synthesized from phenylacetate by the Fris migration reaction [4] . During its synthesis,~30% of 2-HAP is also formed as a by-product along with 4-HAP. Recently,a series of AP based 1-(aryloxypropyl)-4- (chloroaryl) piperazines has been synthesized and studied their atypical antipsychotic activities in mice [5] . Furthermore,4-HAP and its derivatives are used as non-steroidal,anti-inflammatory drugs for the management of dental pain [6] . Several analytical methods,such as gas chromatography [7] ,high performance liquid chromatographic [8] and CE [9] approaches,have been used for the separation and determination of AP,2-HAP,3-HAP and 4-HAP in synthetic mixtures,pharmaceuticals and medicinal plants. However, the reported chromatographic methods required tedious sample preparation,large volumes of organic solvents and lengthy analysis time. Typically,however,a CE method requires a tedious cleaning procedure for the polishing of carbon disc electrode. This notwithstanding,it would be advantageous to establish a simple, rapid,and accurate CE approach for the routine separation of 4- HAP and its isomers in synthetic and in biological samples.
CE is simple,cost-effective and an eco-friendly separation technique for the rapid analysis of drugs in pharmaceutical and biofluids [10] . Moreover,the application of CE has become increasingly widespread for the separation and detection of a wide variety compounds in pharmaceutical and biofluids because it required minimal sample and solvent volumes,short analysis time,and high versatility [10] . Moreover,it holds considerable promise in pharmaceutical research for the accurate analysis of chiral drugs [10, 11] ,neutral compounds [12] and non-ionic compounds [13] . However,it suffers from disadvantages such as low sensitivity,and the short detection pathway of light for the analysis of drugs in pharmaceutical and biological samples. To overcome these problems,several researchers have successfully launched CE with various injection modes,such as head-column ;field-amplified sample stacking [14] ,field amplified polarityswitching sample injection [15] ,and electrokinetic injection [16] which have adequately addressed the sensitivity and short detection pathway problems associated with CE-UV detection methods. However,to date there are no reports on the analysis of AP and its monohydroxy positional isomers by CE with UV detection. The purpose of this study is to develop CE with UV detection for the routine determination of AP and its monohydroxy positional isomers in synthetic and human plasma. Specifically,the use of 4-HAA,as an internal standard,to improve the peak identification and quantification results is also described. 2. Experimental 2.1. Sample preparation
Sodium tetraborate and ammonium acetate buffers (8-20 mmol/L) were prepared by dissolving an appropriate amount of buffer components in deionized water. The buffer pH was adjusted by adding 0.5 mol/L of NaOH. Finally,10 mmol/L of sodium tetraborate (pH 9.5) and acetonitrile (ACN) (5%,v/v) were used in the present study. Stock solutions (AP,2-HPA,3-HAP,4- HAP,and 4-HAA) were prepared in methanol (1.0 mg/mL). These solutions were diluted with 5.0 mmol/L of sodium tetraborate (pH 10.0) for the preparation of standard mixtures. All the prepared samples and buffer were stored in the refrigerator and solutions were filtered with 0.45 μm filter paper before their use in CE.
This study was approved by the Ethics Committee of the Taipe Medical University,Taipe,Taiwan and with the written informed consent of the patients prior to taking plasma samples from the patients (not exposed to any drug for at least 72 h). Four different concentrations of analytes (2.0,4.0,6.0 and 10.0 μg/mL) were spiked into 0.5 mL of plasma and treated with ACN for the precipitation of plasma proteins. The sample vials were vortexed for 3 min and centrifuged at 5000 rpm for 20 min. The resulting clear supernatant solution was evaporated under a gentle nitrogen flow at room temperature. The residue was dissolved in 1.0 mL of MeOH and then analytes were analyzed by CE. The other experimental details,such as chemicals and instrument set-up, were described in Supporting Information. 3. Results and discussion 3.1. Evaluation of separation conditions
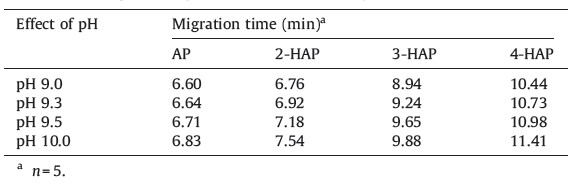
Four analytes pKa values (AP 9.20,2-HAP 8.05,3-HAP 9.25 and 4-HAP 8.05) have already been estimated in the literature [17, 18] . Accordingly,we believe that four analytes are partially dissociated into their anionic forms at pH > 9.0. Therefore,the buffer pH also plays a key role in CE separation because it affects both the charge of the analytes and the strength of the electro-osmotic flow (EOF). Hence,the buffer pH was maintained at 9.5 for the best separation by CE. Fig. 1a and b showed the typical electropherograms of the buffer without analytes and with four analytes. These results revealed that the buffer blank provided a clean base line and the four analytes (AP,2-HAP,3-HAP and 4-HAP) were successfully separated with good resolution. To improve the sensitivity and applicability of the method,we examined the influences of CE separation conditions,such as buffer concentration,buffer pH, organic modifier content,sample matrix,separation voltage and detection wavelength,on the separation of AP and its monohydroxy isomers by CE. The effects of organic modifier volume, sample matrix,separation voltage and analytical data are described in the Supporting Information.

|
Download:
|
| Fig. 1.Electropherogram of buffer without analytes (a) and AP and its monohydroxy isomers (AP,2-HAP,3-HAP and 4-HAP). CE conditions: 10 mmol/L of sodium tetraborate (pH 9.5) and 5% (v/v) ACN (b),10 mmol/L of ammonium acetate (pH 10.0) (c). Injection voltage 10 kV/10 s; separation voltage 15 kV; uncoated fused silica capillary 60 cm, (effective) length ×50 μm i.d; detection wavelength 195 nm. | |
The influence of the buffer concentration on the migration behavior and separation efficiency of four analytes was investigated, since the buffer alters the EOF,zeta-potential,and charge of the analytes,which effects the migration times and the peak responses of analytes. Therefore,two buffers,sodium tetraborate and ammonium acetate (5-20 mmol/L),were selected as buffers to examine the effects of buffer electrolytes on the migration behavior and separation of four analytes (AP - 20.40 μg/mL,2- HAP - 25.30 μg/mL,3-HAP - 20.20 μg/mL and 4-HAP - 18.50 μg/ mL) as shown in Fig. 1b and 1c. We observed that the response currents for the two peaks (AP and 2-HAP) are barely resolved using 10 mmol/L of sodium tetraborate buffer. To improve their resolution,ammonium acetate was used as a buffer (10 mmol/L, pH 9.5) for the best separation of four analytes,however,the ammonium acetate buffer provided entirely different migration behaviors for the four analytes when compared with sodium tetraborate buffer. It was observed that the four analytes migrated within 9.0 min and the two peaks (AP and 2-HAP) and were not well resolved by 10 mmol/L of ammonium acetate buffer (pH 9.0- 10.0). On the other hand,3-HAP peak splitting occurred (Fig. 1c). To avoid the peak splitting,we repeated the CE experiments (n = 5), but identical results were obtained. Interestingly,we noticed that the 3-HAP peak splitting was not observed upon decreasing the 3- HAP concentration from 20.20 μg/mL to 6.06 μg/mL. Since,the peak splitting was reported to depend on experimental conditions and physico-chemical characteristics of analytes and solvents [19, 20] ,we therefore believe that 3-HAP peak splitting is due, probably,to the interaction of the anionic form with ammonium acetate buffer,or incompatibility of buffer pH. Therefore,we selected 10 mmol/L of sodium tetraborate buffer (pH 9.5) for the optimum separation of four analytes by CE. 3.3. Effect of buffer pH
Buffer pH shows significant impact on the electrophoretic mobility of analytes. Since,AP and its monohydroxy isomers possess a keto group and hydroxy group with pKa values in the range 8.05 - 9.25,the analytes are dissociated at pH > 9.5 and the electrophoretic mobility of these analytes are varied in the pH range from 8.10 to 9.30. Therefore,we studied the effect of buffer pH on the migration behavior and separation of four analytes by using buffer pH from 9.0 to 10.0. It was noticed that the two peaks (3-HAP and 4-HAP) were poorly resolved by using buffer pH < 9.20 (<1.5,calculated using Chrom manager software supplied along with the instrument) and the peaks are broadened (peak half width >0.10). Meanwhile,the four analytes are well separated with reasonable migration times using a buffer pH at 10.0. It was also observed that the reproducibility of migration time was decreased due to high current (>78 mA) and low buffer action. Based on their pKa values,we assume that the effective separation can be achieved by using buffer pH above 9.25. As a matter of fact,the four analytes are well separated with reasonable migration times by using sodium tetraborate buffer (pH 9.50) and the obtained data are shown in Table 1.
| Table 1 Effects of buffer pH on migration times of four analytes. |
In order to improve the sensitivity of the present method,a suitable detection wavelength is necessary for the quantification of four analytes by CE. In this study,we used three different wavelengths (195,214 and 254 nm) to measure the absorption efficiency of four analytes with the results shown in Table 2. These results revealed a maximum absorbance at 195 nm for the four analytes and therefore,we selected 195 nm as optimum absorption wavelength for the detection of four analytes.
| Table 2 Observed peak area responses of four analytes and internal standard at different detection wavelengths (nm). |
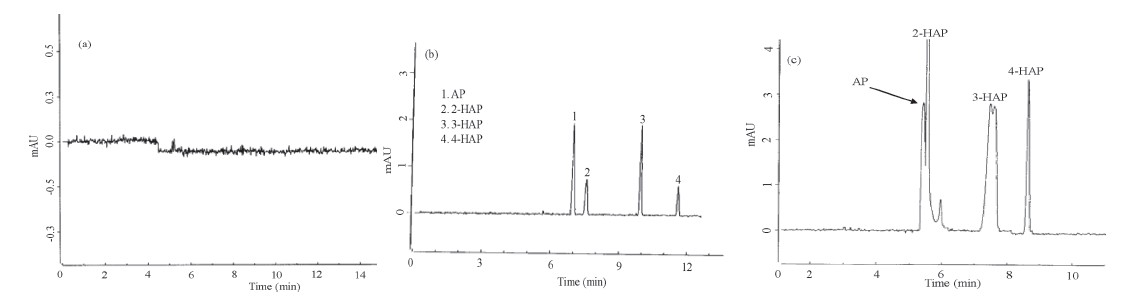
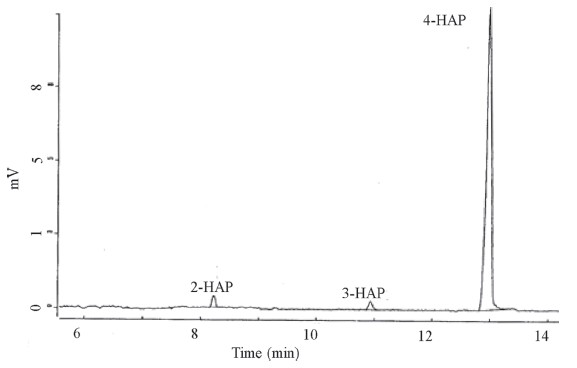
Furthermore,we also studied the applicability of the present method for the trace level separation and determination of 2-HAP, 3-HAP and 4-HAP in synthetic samples. Since,30% of 2-HAP can be obtained as a by-product during the synthesis of 4-HAP,a suitable method is essentially needed for the separation and determination of trace level impurities (2-HAP and 3-HAP) in 4-HAP. Due to absence of 4-HAP in local commercial samples,small amounts of 2- HAP and 3-HAP (0.2%-1.5%) were spiked into 4-HAP and the recoveries calculated by using an external calibration method. The calibration graph was constructed between the known concentrations of 2-HAP and 3-HAP (0.2%-1.5%) and their absorbencies in 4- HAP. The correlation coefficients (R2) were found to be 0.9946 and 0.9921 for 2-HAP and 3-HAP,respectively. The resulting data were shown in Table 3. Based on the above results,the present method successfully separated and determined the trace level impurities (2-HAP and 3-HAP at 0.5%) in 4-HAP as shown in Fig. 2.
| Table 3 Determination of spiked trace-level impurities (2-HAP and 3-HAP) in 4-HAP and analysis of 4-HAP in spiked human plasma sample. |
{kind=link}

|
Download:
|
| Fig. 2.Electropherogram of 0.5% 2-HAP and 3-HAP in 4-HAP. CE conditions are same as shown in Fig. 1. | |
{kind=link}
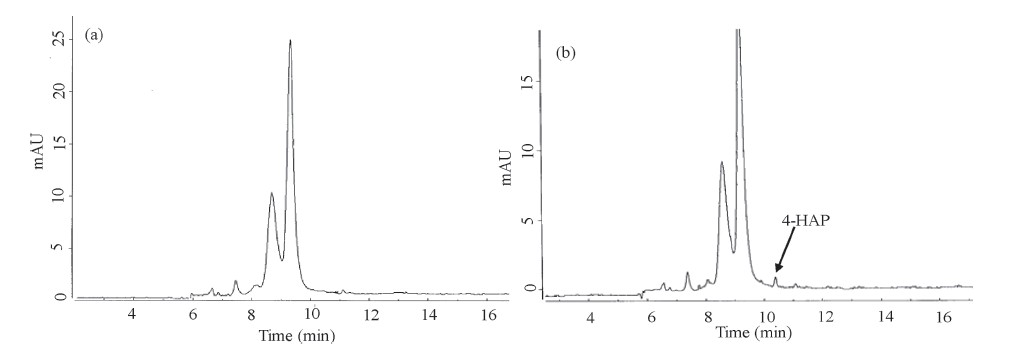
It is well known that 4-HAP has a beneficial effect on acute inflammation and is still under clinical evaluation. Therefore,we investigated the use of our CE method for the determination of 4- HAP in plasma. Briefly,4-HAP (4.0,6.0 and 10.0 μg/mL) was spiked into plasma and detected by the present procedure and obtained results were shown in Table 3. The recoveries were 68.30%-82.0% with a RSD <10.1%. These low recovery values are due to minor interference of biofluids and soluble inorganic species in the plasma. Fig. 3 shows that the electropherograms of plasma sample without 4-HAP (Fig. 3a) and with 4-HAP (4.0 μg/mL) in spiked plasma (Fig. 3b). These results revealed the CE method can effectively separate and detect 4-HAP in plasma samples.

|
Download:
|
| Fig. 3.Electropherogram of plasma (a) without addition of 4-HAP and (b) with spiked 4-HAP (4.0 mg/mL) and CE conditions are same as shown in Fig. 1. | |
{kind=link}
This paper describes a simple and rapid CE method for the separation and determination acetophenone and its monohydroxy isomers (AP,2-HAP,3-HAP and 4-HAP). The four analytes were effectively separated and detected by CE using 10 mmol/L of sodium tetraborate (pH 9.5) with 5% (v/v) ACN. Therefore,the present method is simple and rapid for the separation and quantification of AP and its hydroxy isomers in pharmaceutical industries and in clinical studies.
The authors thank the National Science Council of Taiwan for financial support.
| [1] | The Merck Index, 13th ed., Merck &Co., Inc, Whitehouse Station, NJ, 2002, http:// www.merckbooks.com/mindex/. |
| [2] | A.D. MacKerell Jr., R.S. MacWright, R. Pietruszko, Bromoacetophenone as an affinity reagent for human liver aldehyde dehydrogenase, Biochem. 25(1986) 5182-5189. |
| [3] | Q.W. Zhang, Y.Z. Zhang, Separation and determination of the Cholagogic components in bin hao(Artemisia scoparia) by HPLC, Yaoxue Xuebao 21(1986) 922-927. |
| [4] | B. Gurnule, P.K. Rahangdale, J. Paliwal, R.B. Kharat, Synthesis, characterization and ion-exchange properties of 4-hydroxyacetophenone, biuret and formaldehyde terpolymer resins, React. Funct. Polym. 55(2003) 255-265. |
| [5] | A. Bali, K. Sharma, A. Bhalla, et al., Synthesis, evaluation and computational studies on a series of acetophenone based 1-(aryloxypropyl)-4-(chloroaryl) piperazines as potential atypical antipsychotics, Eur. J. Med. Chem. 45(2010) 2656-2662. |
| [6] | D.R. Mehlisch, The efficacy of combination analgesic therapy in relieving dental pain, J. Am. Dent. Assoc. 133(2002) 861-871. |
| [7] | T.J. Smith, R.H. Wearne, A.F.A. Wallis, Determination of chlorinated benzaldehydes and acetophenones in pulp bleaching effluents by gas chromatography, J. Chromatogr. A 648(1993) 289-293. |
| [8] | Y. Sun, Z. Liu, J. Wang, et al., Determination of four acetophenones in Radix Cynanchi bungei by high performance liquid chromatography-photodiode array detection, Chin. J. Chromatogr. 1(2009) 114-1146. |
| [9] | X. Yao, J. Zhang, G. Chen, Determination of phenolic acetophenones in radix cynanchi paniculati by capillary electrophoresis with electrochemical detection, J. Plant Sci. 2(2007) 273-282. |
| [10] | C. Cruces-Blanco, A.M. Garcia-Campana, Capillary electrophoresis for the analysis of drugs of abuse in biological specimens of forensic interest, Trend. Anal. Chem. 31(2012) 85-95. |
| [11] | X. Liu, S. Li, J. Zhang, X. Chen, Flow injection-capillary electrophoresis frontal analysis method for the study of the interactions of a series of drugs with human serum albumin, J. Chromatogr. B 877(2009) 3144-3150. |
| [12] | T. Wang, J. Tang, W. Wan, S. Zhao, Methyl chloroacetate as an extraction solvent for coupling liquid-liquid semimicroextraction with micellar electrokinetic chromatography through on-capillary decomposition for the separation of neutral compounds with concentration enhancement, J. Chromatogr. A 1147(2007) 105-110. |
| [13] | W. Ding, J.S. Fritz, Separation of nonionic compounds by CE using a lauryl poly(oxyethylene) sulfate additive, Anal. Chem. 69(1997) 1593-1597. |
| [14] | C.X. Zhang, W. Thormann, Head-column field-amplified sample stacking in binary system capillary electrophoresis: a robust approach providing over 1000-fold sensitivity enhancement, Anal. Chem. 68(1996) 2523-2532. |
| [15] | R.L. Chien, D.S. Burgi, Field-amplified polarity-switching sample injection in highperformance capillary electrophoresis, J. Chromatogr. A 559(1991) 153-161. |
| [16] | R. Qurishi, M. Kaulich, C.E. Muller, Fast, efficient capillary electrophoresis method for measuring nucleotide degradation and metabolism, J. Chromatogr. A 952(2002) 275-281. |
| [17] | R.M. Prest, C. Gaspard, E. Laviron, The reduction mechanism of the CO group. Part 2. The electrochemical reduction of p-diacetylbenzene in aqueous medium, J. Electroanal. Chem. 410(1996) 145-154. |
| [18] | B.G. Tehan, E.J. Lloyd, M.G. Wong, et al., Estimation of pKa using semiempirical molecular orbital methods. Part 1: application to phenols and carboxylic acids, Quant. Struct-Act. Relat. 21(2002) 457-472. |
| [19] | S.V. Ermakov, M.Y. Zhukov, L. Capelli, P.G. Righetti, Experimental and theoretical study of artifactual peak splitting in capillary electrophoresis, Anal. Chem. 66(1994) 4034-4042. |
| [20] | C. Rafols, A. Poza, E. Fuguet, M. Roses, E. Bosch, Solute-solvent interactions in micellar electrokinetic chromatography: V. Factors that produce peak splitting, Electrophoresis 23(2002) 2408-2416. |