 2007, 18 (5): 635-644
2007, 18 (5): 635-644


2. 临安大气本底污染监测站, 临安 311307
2. Lin' an Regional Atmospheric Pollution Monitoring Station, Lin' an 311307
尽管大气气溶胶在整个大气中浓度较低, 属于微量成分, 但其在整个气候系统中却起着非常重要的作用。气溶胶粒子通过对太阳辐射和地球长波辐射的散射和吸收, 影响大气的加热/冷却率, 并进而影响整个地气系统的热量收支。此外, 它作为云的凝结和参与云雾的生消, 进而影响云的寿命、微结构、辐射特性和地球大气的水循环[1]。发生在气溶胶颗粒表面上的非均相过程和人类活动排放的废气则作为自然和人为排放气体的汇在大气中的气-粒转换过程中参与大气的化学过程。与气-粒转化过程相反, 大气中也会发生颗粒态固相或液相物质转化成气相的过程。除了通常所见的液体蒸发和固体升华过程外, 大气中某些化学过程也能将粒子转化成气体。例如, 气溶胶粒子中的氯化物可与大气中的酸性物质发生反应生成氯化氢气体, 这是气溶胶对降水酸性影响的重要机制[2]。碳成分的浓度变化对气溶胶辐射强迫以及气溶胶光学厚度的影响是非常明显的[3-5]。另一方面, 气溶胶的寿命相对较短, 其物理化学特性的时空分布很不均匀, 各地的气溶胶特性有各自特性。
目前, 对北京等主要污染城市大气气溶胶特性研究工作开展得较多[6-9], 而对较清洁地区大气气溶胶的特性研究相对较少。自20世纪80年代以来, 我国东部长江三角洲地区经济高速发展, 大量的工业排放和人为活动对该地区环境和区域气候产生了较大的影响[10]。许多工作对该地区大气污染(包括气溶胶)及其对农业生态影响进行了研究[11-16], 但大部分关于气溶胶研究主要是对特定的污染过程以及特定季节的特征进行分析。对粒径分布特点研究也只针对粒径小于10 μm(PM10)和粒径小于2. 5 μm(PM2.5)展开讨论, 还缺少对该地区季节分布变化特点以及粒径分布特点的深入了解。本研究根据2002—2005年“长江三角洲地区大气气溶胶特性研究”、“我国大陆大气本底基准研究”项目在该地区进行的7次观测试验结果, 对该地区气溶胶主要成分(可溶性离子成分和碳成分)以及质量浓度的季节变化及粒径分布特性进行较全面的分析。
1 试验采样点在浙江临安大气本底污染监测站, 该站位于浙江省临安县城北部约10 km处(30°18′N, 119°44′E, 海拔138.6 m)。测点距离浙江省会杭州大约50 km, 其东北大约200 km是上海市, 北部100多公里是苏锡常地区, 西部主要为安徽山区。作为全球大气监测(Global Atmospheric Watch)区域背景监测站, 临安大气本底污染监测站局地污染源较少, 具有较好的区域代表性。采样分春、夏、秋、冬4个季节共进行7次。其中春季为1次采样过程, 自2002年3月30日—4月8日, 其中石英膜3套(每套5张膜), 特氟纶膜6套(每套5张膜), 共计45个样品。夏季为3次采样过程, 分别为2002年8月14—24日, 其中石英膜9套, 特氟纶膜9套; 2003年7月20—30日, 其中石英膜9套, 特氟纶膜9套; 2004年8月17—29日, 其中石英膜9套, 特氟纶膜9套, 夏季共计采样270个。秋季为2次采样过程, 分别为2003年11月7—23日, 其中石英膜10套, 特氟纶膜10套; 2004年11月6—20日, 其中石英膜10套, 特氟纶膜10套, 秋季共计200个样品。冬季2005年1月15日—2月3日为1次采样过程, 其中石英膜8套, 特氟纶膜8套, 共计80个样品。
2 样品的采集、膜处理气溶胶样品是采用改装过的安德森撞击式气溶胶采样器进行收集。改装后的气溶胶采集器可分别采集粒径范围为大于11 μm, 4.7~11 μm, 2.1~4.7 μm, 0.65~2.1 μm, 低于0.65 μm等5级气溶胶颗粒, 改装后的5级气溶胶采样器与改装前的9级采样器相比, 对应尺度段的质量浓度一致性较好[12]。采样膜采用特氟纶和石英膜, 分别做称重分析、离子成分分析和碳成分分析。两种采样膜各装载在两台型号完全相同的采样器上并行采集气溶胶样品。
气溶胶采样膜在采样前后用德国赛多利斯公司十万分之一天平称重, 称重前将采样膜置于温度范围在20±1 ℃, 相对湿度40%±5%条件手套箱内平衡48 h, 反复称量, 称量误差控制在50 μg以内, 取平均值作为采样膜质量。采集到的气溶胶样品编号后立即置于冰箱中保存。为了尽量避免吸附于石英膜上的有机成分流失, 运送过程中将采样膜置于-20 ℃保温箱中保存。
为确保采样流量的准确性, 本工作采用通过国家计量院鉴定的干空气表, 在每次采样前和采样结束后对采样器流量进行检查。
气溶胶离子成分在中国气象科学研究院大气化学实验室用Dionex 500离子色谱仪(IC)和HITACHI 180-70原子吸收(AAS)分析。采样膜处理方法如下:取半张采满气溶胶的特氟纶膜, 剪成碎片后浸入25 ml去离子水, 用超声振荡器萃取2~4 h, 各取清液10 ml用离子色谱仪(IC)和原子吸收光谱仪(AAS)分析。本文分析离子包括:氟离子(F-), 氯离子(Cl-), 硝酸根离子(NO3-), 硫酸根离子(SO42-), 氨根离子(NH4+), 钾离子(K+), 钠离子(Na+), 钙离子(Ca2+), 镁离子(Mg2+)等9种离子。EC/OC在北京大学环境中心实验室用Sunset热-光碳分析仪分析(Sunset Lab实验室, 美国), 采样前石英膜在600 ℃高温焙烧4 h以上, 石英膜采样前后保存在-20 ℃环境中。
3 结果分析 3.1 试验期主要气候背景2002年3—4月平均气温达12.7 ℃, 比常年平均偏高3.4 ℃, 4月中旬前期达最高值。2002年夏季平均气温26.0 ℃, 6月下旬开始至8月中旬因出现连续阴雨天气, 旬气温持续比常年偏低, 8月中旬气温仅为23.6 ℃; 2003年7—8月气温明显偏高, 6月30日—8月10日出现持续晴热高温天气, 日最高气温≥35 ℃的日数占79%(33/42), 特别是7月19日—8月5日的最高气温≥37 ℃, 其中不低于39 ℃的日数占61%(11/18), 出现历史罕见的高温天气; 2004年7月16日—8月11日连续出现高温天气, 但采样期间气温接近常年水平, 平均气温27.6 ℃。2003年11月上中旬的降温幅度最大, 从上旬的16.0 ℃降到中旬的11.5 ℃, 2004年采样期间平均温度达13.4 ℃。2005年1月和2月平均气温为2.7 ℃。
3.2 质量浓度季节变化特性通过对该地区不同尺度段气溶胶质量浓度的测量, 计算出整个尺度范围内气溶胶质量浓度, 并对其在不同季节变化特点进行了分析。7次采样过程的质量浓度数据列于表 1中, 质量浓度数据在2002年夏季的一次实验中没有对气溶胶采样膜进行称重, 故该数据缺测。结果表明, 夏季的两次采样过程气溶胶质量浓度实际差别为8 μg/m3, 平均质量浓度为65.7 μg/m3, 在四季中为最低。秋季两次采样过程气溶胶质量浓度分别为99.7 μg/m3和95.3 μg/m3其变化幅度不超过5 μg/m3, 平均值为98.7 μg/m3, 高于夏季数值。春季大气气溶胶质量浓度最高, 达到534 μg/m3, 主要是由于春季采样过程中包括一次沙尘暴过程, 冬季次之, 质量浓度117.21 μg/m3。四季气溶胶平均质量浓度特点见图 1。总体来讲, 该地区气溶胶质量浓度年平均水平与污染城市比较偏低, Zhang等[17]研究表明, 西安年平均气溶胶质量浓度为400 μg/m3。
|
|
表 1 7次采样过程质量浓度(单位: μg/m3) Table 1 The mass concentration of 7 campaigns(unit: μg/m3) |


|
|
| 图 1. 气溶胶质量浓度年度变化(单位: μg/m3) Fig 1. The yearly variety of aerosol mass concentration(unit: μg/m3) | |
夏季气溶胶质量浓度最低的主要原因是夏季地面扬尘贡献较小, 同时正值梅雨季节, 降水较多, 大气颗粒物大部分被冲刷稀释到地面, 对大气具有净化作用。冬季气溶胶浓度高, 是夏季气溶胶浓度的2倍, 原因在于冬季空气相对干燥, 加之逆温层较强较厚, 不利于污染物的扩散, 延长气溶胶在空中停留时间。冬秋两季浓度无明显差别, 说明冬季采暖排放对气溶胶贡献不大。
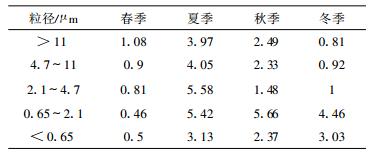
3.3 气溶胶质量浓度尺度分布一般而言, 四季中气溶胶质量浓度尺度分布总体特点相同, 大部分颗粒物均分布在粒径小于11 μm(PM11)范围内, 超过一半的粒子分布在小于2.1 μm(PM2.1)范围内。PM11质量浓度年平均值189.3 μg/m3, 春季为500 μg/m3(与沙尘天气有关), 夏季为59.7 μg/m3, 秋季为92.4 μg/m3, 冬季为104.9 μg/m3, 分别占气溶胶总质量浓度的93.6%, 90.3%, 94.8%和89.5%。其质量浓度水平与长江三角洲地区1999年秋季PM10质量浓度(98 μg/m3)[12]接近, 而与北京1994年冬季PM10的质量浓度(555.6 μg/m3)[10]以及北京城区PM10小时平均值400 μg/m3[18-19]低很多, 主要原因是北京冬季燃煤大量排放所致。临安PM2.1质量浓度年平均值为114.6 μg/m3, 其中, 春季为280.2 μg/m3, 夏季为41.8 μg/m3, 秋季为64.8 μg/m3, 冬季为71.6 μg/m3, 分别占气溶胶总质量浓度52.5%, 63.6%, 66.4%和61.1%。秋季PM2.1与PM2.5浓度(90 μg/m3)[12]比较偏低, 与污染城市比较PM2.5质量浓度也偏低。例如, 武汉PM2.5质量浓度平均值为139 μg/m3[20], 大同PM2.1平均值为130 μg/m3[21], 北京1999年6月PM2.5质量浓度为136 μg/m3[22]。按照美国PM2.5国际标准要求, 24 h PM2.5质量浓度平均不得超过65 μg/m3, 该地区只有夏季和秋季质量浓度符合标准。不同季度细粒子分布特征表现有所不同, 夏季在小于0.65 μm尺度上气溶胶质量浓度最大。而在春、秋、冬季最大值出现在0.65~2.1 μm尺度范围内。主要原因除与扬尘对积聚态大气气溶胶贡献外, 还与气溶胶生成与长大有关。一般而言, 粒径 < 0.05 μm气溶胶粒子被称为爱根核模, 主要来源于燃烧过程产生的一次气溶胶粒子和气体分子通过化学反应均相成核转换成的二次气溶胶粒子。因为它的粒径小, 数量多, 表面积总量大, 随着时间的推移, 易由小粒子的相互碰撞而合并成大粒子[23]。春、秋、冬季空气相对夏季而言较干燥, 新生成的气溶胶在大气中停留时间较长, 有助于气溶胶在大气中的碰并与增长, 从而导致积聚态气溶胶浓度增加。而夏季空气湿度较大, 加之降水过程频繁, 细颗粒来不及长大便随降水离开大气降落至地面进而消失(图 2)。

|
|
| 图 2. 不同粒径气溶胶质量浓度年度变化 Fig 2. The yearly variety of aerosol mass concentration in different size stage | |
3.4 可溶性离子成分季节变化
全谱气溶胶可溶性离子浓度季节变化特点与气溶胶质量浓度季节变化特点相同。春季最高达60.3 μg/m3, 夏季最低为32.3 μg/m3, 秋季为41.1 μg/m3, 冬季为42.9 μg/m3。影响可溶性离子成分浓度的因素很多, 其中温度、湿度的影响尤为突出, 由于2002年夏季连续阴雨天气以及低温影响, 导致2002年可溶性离子浓度偏高, 达51.7 μg/m3, 2003年和2004年可溶性离子浓度分别为17.08 μg/m3和28.45 μg/m3, 尽管2002年可溶性离子浓度偏高, 但夏季可溶性离子浓度平均值低于其他季节的事实是存在的。而且夏季的离子成分在气溶胶中占的比例也较大。可溶性离子占气溶胶质量浓度的百分比四季中有所不同, 春季为11.3%, 夏季为49.4%, 秋季为42.1%, 冬季为36.6%。可见, 可溶性离子成分在气溶胶中占的比例很大, 夏季可溶性离子约占气溶胶浓度的一半。
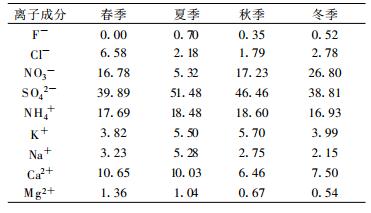
不同离子成分变化特点显示, SO42-, NH4+和NO3-在可溶性离子成分中均占有主导地位。3种离子浓度之和约占可溶性离子总量的75%~83%。其中SO42-在16.6~24.0 μg/m3之间, 占离子总量百分比春季为39.9%, 夏季为51.5%, 秋季为46.5%和冬季为38.8%(表 2), NH4+质量浓度为6.0~10.6 μg/m3, 占离子总量百分比分别为春季17.7%, 夏季18.5%, 秋季18.6%和冬季16.9%; NO3-质量浓度为1.7~11.5 μg/m3, 占离子总量的百分比春季为16.8%, 夏季为5.3%, 秋季为17.2%和冬季为26.8%。从以上分析可见, SO42-在可溶性离子成分中扮演着重要角色。NH4+所占比例较稳定, 大气气溶胶中NH4+的唯一重要来源是气态NH3的凝结[24], 说明该地区气态NH3的来源相对稳定。NO3-在夏季浓度明显低于春、秋、冬季, 这一方面与硝酸盐本身的高温不稳定性有直接关系, 一般硝酸盐在温度达到30 ℃以上时, 其稳定性极差, 很容易挥发转化成气态NO2[25]; 另一方面, 采样用的特氟纶膜对硝酸铵的损失也与采样时的气象条件(温度、湿度)有很大的关系, 当温度超过25 ℃时, 特氟纶膜对挥发性硝酸铵的损失可达百分之百[26]。采样期间的气象数据表明, 临安地区2003年7月下旬平均气温达31.4 ℃, 创历史同期最高记录, 比原最高记录2001年31.1 ℃高0.3 ℃。夏季高温天气是导致所观测的气溶胶中硝酸盐浓度降低的主要原因。而SO42-在夏季贡献略高于其他3个季节, 因为大部分硫酸盐来源于二次气溶胶, SO42-是气粒转化过程主要产物, 夏季高温天气, 有利于发生光化学反应, 促进气-粒转化过程发生。
|
|
表 2 不同季节各种离子成分占总离子成分百分比(单位: %) Table 2 The percentage of different ions in different seasons(unit: %) |
3.5 可溶性离子平衡与相关性分析
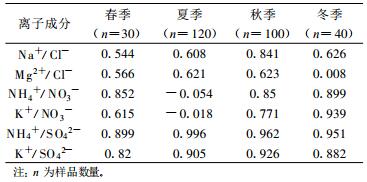
进一步分析表明, 四季中阳离子当量浓度均高于阴离子当量浓度, 且阳离子与阴离子当量浓度的比值在1.16~1.66之间波动。其主要原因可能与一些阴离子, 如碳酸根离子CO32-、碳酸氢根离子HCO3-和有机酸根离子没有分析有关。在对离子进行相关性分析时, 采用Pearson相关性系数来判断[27]。表 3列出各离子在α=0.01水平上显著相关物种间的相关系数。显著相关物种相关系数越大, 说明两种离子形成稳定化合物的可能性越大, 其在大气中的变化形式越相似, 或者来源越相似。NH4+与SO42-在四季中相关性系数均在0.9以上, K+与SO42-相关系数也在0.8以上。Na+与Cl-相关系数在0.54~0.84之间, NH4+与NO3-除夏季显著不相关外, 其余3个季节相关系数均在0.8以上, 显著相关。由此可见, 气溶胶以(NH4)2SO4, K2SO4, NaCl, MgCl2, NH4NO3, KNO3等化合物形式存在的可能性极大。夏季NH4+, K+与NO3-的相关系数极低, 而NH4+与SO42-的相关系数明显增加, 如上文所述, 由于夏季SO42-高温挥发现象出现, 导致夏季NO3-浓度明显降低, 因此, 阳离子在形成化合物时, 只能首选SO42-与其合成稳定化合物, 南方夏季高温天气抑制了硝酸盐类气溶胶的形成。
|
|
表 3 四季气溶胶可溶性离子成分相关系数 Table 3 The correlation between ions in the 4 seasons |
3.6 可溶性离子尺度分布特性
离子成分在5个尺度范围内的浓度分布特点表现为:春、冬两季在0.65~2.1 μm范围内3种离子浓度都不同程度的高于粒径 < 0.65 μm范围内的离子浓度。在大于2.1 μm粒径范围内标志着土壤扬尘的代表性离子Ca2+浓度以及海盐溅沫Na+, Cl-浓度相对于小于2.1 μm粒子而言均较高(图 3), 说明土壤源, 海盐主要分布于粗粒子中。随着颗粒越来越细, 离子成分分布越来越集中, 最终集中于SO42-, NH4+, NO3-离子成分上, 说明硫酸盐、硝酸盐和铵盐集中分布在细颗粒中。PM2.1范围SO42-浓度春季为18.1 μg/m3, 夏季为16.2 μg/m3, 秋季为17.3 μg/m3, 冬季为15.3 μg/m 3, 比文献[12]长江三角洲地区PM 2.5中SO42-在1999年秋季浓度水平(21.2 μg/m3)低, 且其浓度季节变化不大, NH4+季节变化特点与SO42-相似, 浓度变化幅度较小, 由夏季最小值6.1 μg/m3到春季最大值的8.8 μg/m3。NO3-在PM2.1中浓度随温度变化较明显, 春季为5.6 μg/m3, 夏季为0.5 μg/m3, 秋季为4.8 μg/m3, 冬季为8.8 μg/m3, 温度越低, 其浓度越高。

|
|
| 图 3. 不同尺度气溶胶离子浓度季节特征 Fig 3. The seasonal characteristic of aerosol ionic concentration in different size stage | |
3.7 不同尺度阴阳离子平衡及相关分析
分析不同尺度阴阳离子平衡关系, 结果发现阴阳离子当量浓度比值在夏季和冬季随尺度变化幅度较大。夏季细粒子(粒径 < 2.1 μm)当量浓度比值为1.32, 粗粒子(粒径>2.1 μm)当量浓度比值为3.59, 冬季细粒子当量浓度比值为0.99, 粗粒子当量浓度比值为3.74, 春、秋两季阴阳离子当量浓度比值分别在1.02~2.08和1.19~2.47之间波动。说明颗粒越粗, 阴离子中未知成分的浓度越高, 而在粗粒子中, Ca2+浓度占有相当一部分比例, 气溶胶中的钙离子Ca2+与碳酸根离子CO32-或碳酸氢根离子HCO3-化合成碳酸钙CaCO3, 碳酸氢钙Ca(HCO3)2的可能性很大, 同时也可以证明未知成分为CO32-, HCO3-的可能性。分析粗粒子与细粒子中各离子间相关性(表 4)。表中数值表示两物种在α=0.01水平上的相关系数, 括号中数值表明两物种在α=0.01水平上不显著相关, 黑体数值表明两物种在α=0.01水平上强显著相关。总体而言, 粗粒子中Na+与Cl-相关性系数均在0.7以上, 说明海盐溅沫产生的气溶胶大部分分布在粗粒子中, Ca2+与Mg2+, Na+显著相关, NH4+, Mg2+与Cl-显著相关, 显现出土壤类扬尘的化学组分特点。春季细粒子中NH4+与SO42-, NO3-在α=0.01水平上显著相关, NH4+与(SO42- +NO3-)当量浓度比值为1:1.05, 可以判断出春季细粒子气溶胶中存在NH4NO3,(NH4)2SO4。夏季细粒子中NH4+与SO42-显著相关, 当量浓度比值为1:1.02, 秋季细粒子中NH4+, K+与SO42-, NO3-强显著相关, (NH4+ +K+)与(SO42- +NO3-)当量浓度比值为1:1.05, 冬季细粒子与秋季相似, NH4+, K+与SO42-, NO3-强显著相关,(NH4+ +K+)与(SO42- +NO3-)当量浓度比值为1:0.95, 由此可见, 细粒子中, 除夏季主要以硫酸氨(NH4)2SO4形式存在外, 其余3季气溶胶中主要以硫酸氨(NH4)2SO4, 硝酸铵NH4NO3, 硫酸钾K2SO4, 硝酸钾KNO3形式存在。
|
|
表 4 四季粗细粒子气溶胶中各离子成分相关分析 Table 4 Correlation coefficients of different ions in four seasons |

3.8 全谱碳浓度及尺度变化特征
大气中元素碳、颗粒态有机碳是气溶胶对辐射过程影响最大的组分, 尤其是元素碳具有很强的吸收作用, 其主要由一次气溶胶如化石燃料不完全燃烧所产生的, 有机碳化合物则包括污染源直接排放的一次有机碳粒子和气态碳氢化合物通过光化学反应等途径生成二次有机碳化合物[28-32]。为此, 本文对临安地区气溶胶中元素碳及有机碳成分分布特点作了详细分析。首先分析了气溶胶全谱范围内有机碳、元素碳浓度季节变化特点(图 4), 元素碳在春季浓度异常高, 是其余3个季节元素碳浓度的5倍。主要是因为春季采样包括一次沙尘暴过程, 而元素碳大多数来源于一次气溶胶, 沙尘天气的出现, 将地面一次气溶胶向大气中输送, 从而使得春季气溶胶中元素碳浓度明显偏高。夏季元素碳(EC)质量浓度最低为4.7 μg/m3, 秋冬季EC浓度均有所增加。有机碳质量浓度在21.4~15.1 μg/m3范围内波动, 其中春季最高, 随着季节变化, 逐渐降低, 冬季达最小值15.1 μg/m3。该地区元素碳(EC)、有机碳(OC)质量浓度与其他城市地区EC, OC浓度相比有高有低。例如北京7—8月OC质量浓度为12 μg/m3, EC为5.4 μg/m3[33], 上海秋季OC质量浓度为29 μg/m3, EC为12 μg/m3, 冬季OC质量浓度为25 μg/m3, EC为10 μg/m3[34], 广州1—2月OC质量浓度为23 μg/m3, EC为8.3 μg/m3[35], 中国香港市区1—2月OC质量浓度为9.6 μg/m3, EC为4.7 μg/m3。

|
|
| 图 4. 碳成分全谱年度变化 Fig 4. The yearly variety of carbon concentration | |
为了更加深入的研究EC, OC形成气溶胶的特性, 本文对不同粒径范围内EC, OC浓度分布特点进行了分析(如图 5所示)。结果显示, 随着气溶胶粒子尺寸的减少, 有机碳、元素碳浓度不断增加。秋冬两季有机碳在0.65~2.1 μm范围内出现极大值, 春夏两季小于0.65 μm气溶胶中OC浓度最高。另外, 粒径小于2.1 μm气溶胶中有机碳的浓度明显高于粒径大于2.1 μm气溶胶中有机碳的浓度, 夏季约为2倍, 秋季约为3倍, 冬季约为6倍。同样, 细粒子中元素碳浓度也高于粗粒子中元素碳的浓度。

|
|
| 图 5. 有机碳(a)和元素碳(b)尺度分布年度变化 Fig 5. The yearly size kistrbutions of organic carbon(a)and element carbon(b) | |
EC与OC比值可以用来评价一次气溶胶与二次气溶胶对大气气溶胶的贡献。一般而言, 如果大气气溶胶中OC与EC比值低, 说明气溶胶中二次有机碳浓度低, 大部分为一次含碳化合物[36]。相反, 如果气溶胶中OC与EC比值高于2, 就可以判断二次有机气溶胶在大气气溶胶中占有很大比例[37-38]。临安地区气溶胶中OC与EC比值在不同季节以及不同粒径范围内均存在着明显的差异(表 5)。表 5表明, 春季气溶胶5个粒径尺度范围内OC与EC比值均较低, 说明一次气溶胶占有绝对优势。冬季粒径>2.1 μm气溶胶颗粒中, OC与EC比值均小于2, 粒径 < 2.1 μm颗粒中OC与EC比值大于2, 说明冬季粗颗粒以一次气溶胶为主, 细颗粒则有一定数量的二次气溶胶存在。夏秋两季气溶胶在5个尺度范围内OC与EC比值几乎均大于2, 说明夏秋两季气溶胶中含碳化合物中含有一定的二次有机气溶胶。
|
|
表 5 四季不同尺度气溶胶OC与EC比值比较 Table 5 The seasonal ratio of OC to EC comparison in different size stages |
另外值得注意的是, K+与OC, EC之间存在较好的相关性。一般情况下, 钾来源于生物质的燃烧[39], 临安地区春、夏、秋、冬季OC与K+相关系数分别为0.62, 0.72, 0.85, 0.82。EC与K+春、夏、秋、冬季相关系数分别为-0.58, 0.50, 0.56, 0.59。除春季EC与K+成负相关外(春季K+可能主要来自土壤等粉尘来源), 其余结果表明, 临安地区气溶胶中含碳化合物来源与生物质燃烧有关。
4 结论1) 临安地区全谱大气气溶胶全年平均质量浓度范围为65.7~534 μg/m3。其中春季质量浓度达最大值。离子浓度变化范围32.3~60.3 μg/m3, 春季浓度最高, 夏季浓度最低, 其中SO42-, NH4+, NO3-在离子成分中占有主导地位。OC, EC浓度春季最高, 冬季最低。
2) 从气溶胶粒径分布特征看, 90%左右气溶胶分布在粒径 < 11 μm的颗粒中, 粒径 < 2.1 μm气溶胶约占气溶胶总质量浓度的52%以上。可溶性离子浓度尺度变化特征明显, 细粒子中可溶性离子成分浓度高于粗粒子中可溶性离子浓度。
3) 从不同粒径范围的组成看, 粗粒子主要由土壤类扬尘以及海盐溅沫产生的气溶胶组成。细粒子中离子成分集中于SO42-, NH4+和NO3-离子上。细粒子中, 除夏季主要以(NH4)2SO4形式存在外, 其余3个季节气溶胶中主要以(NH4)2SO4, NH4NO3, K2SO4, KNO3形式存在。
4) 碳成分粒径分布特点为颗粒越细, 碳成分浓度越高, 夏秋两季以二次有机气溶胶为主, 春季以一次气溶胶为主, 冬季粗粒子以一次气溶胶为主, 细粒子中以二次气溶胶为主。
致谢 感谢临安大气本底污染监测站技术人员在现场观测中的大力协助。| [1] | 许黎, 冈田菊夫, 张鹏, 等. 北京地区春末-秋初气溶胶理化特性的观测研究. 大气科学, 2002, 26, (3): 401–411. |
| [2] | 王明星. 大气化学. 北京: 气象出版社, 1991. |
| [3] | IPCC, Climate Change 2001:The Scientific Basis. Cambridge: Cambridge University Press, 2001: 1-881. |
| [4] | Surabi Menon, James Hansen, Larissa Nazarenko, et al. Climate effects of black carbon aerosols in China and India. Science, 2002, 297: 2250–2253. DOI:10.1126/science.1075159 |
| [5] | Jacobson M Z, Strong radiative heating due to the mixing state of black carbon in atmospheric aerosols. Nature, 2001, 409: 695–697. DOI:10.1038/35055518 |
| [6] | Jacobson M Z, Control of fossil-fuel particulate black carbon plus organic matter, possibly the most effective method of slowing global warming. J Geophys Res, 2002, 107, (D19): 4410. DOI:10.1029/2001JD001376 |
| [7] | 杨复沫, 贺克斌, 马永亮, 等. 北京大气PM2.5中微量元素的浓度变化特征与来源. 环境化学, 2003, 24, (6): 33–37. |
| [8] | 杨复沫, 贺克斌, 马永亮, 等. 北京PM2.5化学物种的质量平衡特征. 环境化学, 2004, 23, (3): 326–333. |
| [9] | Shi Zongbo, Shao Longyi, Jones T P, et al. Characterization of airborne individual particles collected in an urban area, a satellite city and a clean air area in Beijing, 2001. Atmos Environ, 2003, 37: 4097–4108. DOI:10.1016/S1352-2310(03)00531-4 |
| [10] | 周福民, 孙庆瑞, 王美蓉, 等. 北京中关村地区气溶胶的酸性测量. 环境科学, 1998, 19, (2): 6–11. |
| [11] | 周秀骥. 长江三角洲地层大气与生态系统相互作用研究. 北京: 气象出版社, 2004. |
| [12] | Xu J, Bergin M H, Yu X, et al. Measurement of aerosol chemical, physical and radiative properties in the Yangtze delta region of China. Atmos Environ, 2002, 36: 161–173. DOI:10.1016/S1352-2310(01)00455-1 |
| [13] | 杨东贞, 于晓岚. 临安本底站微量气体浓度分布特征及其对气溶胶的影响. 应用气象学报, 1995, 6, (4): 400–406. |
| [14] | 杨东贞, 颜鹏, 张养梅, 等. WMO区域本底站气溶胶特征分析. 第四纪研究, 2006, 26, (5): 733–741. |
| [15] | 颜鹏, 张养梅, 王淑凤, 等. 临安夏季气溶胶离子成分尺度分布特征. 气象学报, 2005, 63, (6): 980–987. |
| [16] | 颜鹏, 杨东贞, 汤洁, 等. 临安一次沙尘暴过程影响气溶胶物理化学特性演变的初步分析. 第四纪研究, 2004, 24, (4): 437–446. |
| [17] | Zhang X Y, Cao J J, Li L M, et al. Characterization of atmospheric aerosol over Xi'an in the south margin of the Loess Plateau, China. Atmos Environ, 2002, 36: 4189–4199. DOI:10.1016/S1352-2310(02)00347-3 |
| [18] | Ando M, Katagiri K, Tamura K, et al. Indoor and outdoor air pollution in Tokyo and Beijing supercities. Atmos Environ, 1994, 30: 695–702. |
| [19] | 时宗波, 邵龙义, 李红, 等. 北京市西北城区取暖期环境大气中PM10的物理化学特征. 环境科学, 2002, 23, (1): 31–34. |
| [20] | Waldman J M, Lioy P J, Zelenka M, et al. Winter time measurements of aerosol acidity and trace elements in Wuhan, a city in central China. Atmos Environ, 1991, 25B: 113–120. |
| [21] | Salmon L G, Christoforou C S, Cass G R, et al. Air pollutants in the Buddhist cave temples at the Yungang Grottoes, China. Environ Sci Technol, 1994, 28: 805–811. DOI:10.1021/es00054a010 |
| [22] | Bergin M H, Cass G, Xu J, et al. Aerosol reactive, physical and chemical properties in Beijing during June 1999. J Geophysical Res, 2001, 106, (D16): 17969–17980. DOI:10.1029/2001JD900073 |
| [23] | 唐孝炎. 大气环境化学. 北京: 高等教育出版社, 2002. |
| [24] | Admas P J, Seinfeld J H, Koch D M, et al. Global concentrations of troposphere sulfate, nitrate and ammonium aerosol simulated in a general circulation model. J Geophys Res, 1999, 104: 13791–13823. DOI:10.1029/1999JD900083 |
| [25] | 刘振海. 分析化学手册. 北京: 高等教育出版社, 2000. |
| [26] | Schaap M, Spindler G, Schulz M, et al. Artefacts in the sampling of nitrate studied in the "INTERCOMP" campaigns of EUROTRAC-aerosol. Atmos Environ, 2004, 38: 6487–6496. DOI:10.1016/j.atmosenv.2004.08.026 |
| [27] | 数学手册编写组. 数学手册. 北京: 高等教育出版社, 1998. |
| [28] | Turpin B J, Huntzicker J J, Identification of secondary organic aerosol episodes and quantitation of primary and secondary organic aerosol concentrations during SCAQS. Atmos Environ, 1995, 29: 3527–3544. DOI:10.1016/1352-2310(94)00276-Q |
| [29] | Turpin B J, Lim H J, Species contribution to PM2.5 concentrations:Revising common assumption for estimating organic mass. Aerosol Sci Technol, 2001, 35: 602–610. DOI:10.1080/02786820119445 |
| [30] | Turpin B J, Huntzicker J J, Larson S M, et al. Los Angeles summer midday particulate carbon:Primary and secondary aerosol. Environ Sci Technol, 1991, 25: 1788–1793. DOI:10.1021/es00022a017 |
| [31] | James D A, Alfarra M R, Keith N B, et al. Quantitative sampling using an aerodyne aerosol mass spectrometer 2:Measurements of fine particulate chemical composition in two U K cities. J Geophys Res, 2003, 108, (D3): 4091. DOI:10.1029/2002JD002359 |
| [32] | 孙宏, 张择, 裴力民, 等. 齐齐哈尔市大气气溶胶中有机碳和元素碳污染初步分析. 齐齐哈尔师范学院学报(自然科学版), 1997, 17, (2): 57–61. |
| [33] | He Z, Kim Y J, Ogunjobi K O, et al. Carbonaceous aerosol characteristic of PM2.5 particles in northeastern Asia in summer 2002. Atmos Environ, 2004, 38: 1795–1800. DOI:10.1016/j.atmosenv.2003.12.023 |
| [34] | Ye B, Ji X, Yang H, et al. Concentration and chemical composition of PM2.5 in Shanghai for a 1-year period. Atmos Environ, 2003, 37: 499–510. DOI:10.1016/S1352-2310(02)00918-4 |
| [35] | Cao J J, Lee S C, Ho K F, et al. Characteristics of carbonaceous aerosol in Pearl River Delta Region China during 2001 winter period. Atmos Environ, 2003, 37: 1451–1460. DOI:10.1016/S1352-2310(02)01002-6 |
| [36] | Zhang X Y, Wang Y Q, Wang D, et al. Charaterization and sources of regional-scale transported carbonaceous and dust aerosol from different pathways in coarstal and sandy land of China. J Geophys Res, 2005, 110: D15301. DOI:10.1029/2004JD005457 |
| [37] | Chow J C, Watson J G, Lu Z, et al. Descriptive analysis of PM2.5 and PM10 at regionally representative locations during SJVAQS/AUSPEX. Atmos Environ, 1996, 30: 2079–2112. DOI:10.1016/1352-2310(95)00402-5 |
| [38] | Gray H A, Cass G R, Huntzicker J J, Characteristics of atmospheric organic and elemental carbon particle concentrations in Los Angeles. Environ Sci Technol, 1986, 20: 580–589. DOI:10.1021/es00148a006 |
| [39] | Andreae M O, Soot carbon and excess fine potassium:Long range transport of combustion-derived aerosol. Science, 1983, 220: 1148–1151. DOI:10.1126/science.220.4602.1148 |