 2018, Vol. 38
2018, Vol. 38 

甲苯磺酸索拉非尼是德国拜耳公司研制的一种新型抗癌药物,最早于2005年12月20日在美国获准上市,目前已在世界50多个国家被批准用于治疗晚期肾细胞癌,本品已于2006年9月12日获国家食品药品监督管理局批准进口中国[1-6]。目前已有较多文献报道甲苯磺酸索拉非尼的含量测定方法,对于甲苯磺酸索拉非尼原料药的有关物质方法还鲜有研究。本研究建立了HPLC法测定甲苯磺酸索拉非尼中有关物质,并根据ICH等相关指导原则进行了方法学验证,结果方法专属、精密、准确。
1 仪器与试药 1.1 仪器Thermo Fisher U3000型高效液相色谱仪(戴安仪器公司),Waters SBride C18色谱柱(250 mm×4.6 mm,5 μm;填料:十八烷基硅烷键合硅胶;Waters公司);MS-105DU型十万分之一分析天平(梅特勒公司);HT-300BQ型超声清洗器(功率300 W、频率40 kHz,济宁恒通超声电子设备有限公司)。
1.2 试药甲苯磺酸索拉非尼原料(自制,批号为20150119-SL、20150302-SL、20150311-SL);甲苯磺酸索拉非尼工作对照品(自制,纯度为99.8%,水分0.21%,批号为20150204-DZ);杂质Y-1对照品(自制,纯度为99.99%,批号为20150128-Y1);杂质Y-2对照品(自制,纯度为99.17%,批号为20150301-Y2);杂质Y-3对照品(自制,纯度为99.54%,批号为20150301-Y3);杂质Y-4对照品(自制,纯度为99.92%,批号为20150120-Y4);杂质M1对照品(自制,纯度为99.57%,批号为20150405-M1);杂质A对照品(深圳市斯坦德化工科技有限公司,批号152941S-TZ-01,纯度为99.53%);杂质D对照品(深圳市斯坦德化工科技有限公司,批号152944S-TZ-01,纯度为98.31%);杂质F对照品(深圳市斯坦德化工科技有限公司,批号152946S-TZ-01,纯度为100.00%);杂质G对照品(深圳市斯坦德化工科技有限公司,批号152947S-TZ-01,纯度为100.00%);杂质K对照品(TLC,批号1674-006A2,纯度为99.7%);杂质N氧化物对照品(TLC,批号1477-070A4,纯度为99.8%)。
乙腈(色谱纯,Fisher),乙醇(色谱纯,Fisher),磷酸二氢钾(色谱纯,MREDA),磷酸(色谱纯,MREDA),水为自制纯化水。
2 方法与结果 2.1 色谱条件色谱柱为Waters C18柱(250 mm×4.6 mm,5 μm),流动相A为pH 2.4磷酸盐缓冲液-乙腈-乙醇(90:6:4),流动相B为pH 2.4磷酸盐缓冲液-乙腈-乙醇(20:48:32),进行梯度洗脱,流速为1.0 mL·min-1,柱温为35 ℃,检测波长为235 nm,进样量为5 μL。梯度洗脱见表 1。
|
|
表 1 梯度洗脱程序 Table 1 The program of gradient elution |
取甲苯磺酸索拉非尼原料约37.5 mg,精密称定,置25 mL量瓶中,用稀释剂[流动相A-B(1:3)]溶解并稀释至刻度,摇匀,即得。
2.2.2 杂质A、D、F、M1混合溶液取杂质A、D、F、M1各约10 mg,精密称定,分别置10 mL量瓶中,用稀释剂溶解并稀释至刻度,摇匀,精密量取上述各溶液3 mL置25 mL量瓶中,用稀释剂稀释至刻度,摇匀,再精密量取该溶液9 mL置200 mL量瓶中,用稀释剂稀释至刻度,摇匀,即得混合杂质溶液。
2.2.3 对照溶液精密量取供试品溶液1 mL,置100 mL量瓶中,用稀释剂稀释至刻度,摇匀,即得对照溶液。
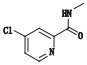
2.3 杂质分析索拉非尼由对氨基苯酚和4-氯-N-甲基吡啶-2-甲酰胺为起始原料,经过3步反应得到甲苯磺酸索拉非尼,其合成反应步骤如图 1。

|
图 1 甲苯磺酸索拉非尼的合成反应步骤 Figure 1 Synthesis reaction step of sorafenib tosylate |
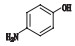
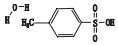
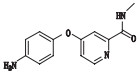
根据其合成路线,推测其可能的杂质及各杂质名称与结构见表 2。
|
|
表 2 杂质 Table 2 Impurities |
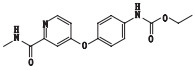
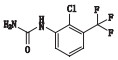
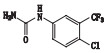
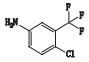
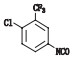
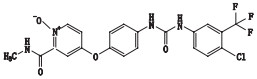
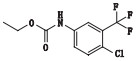
取杂质Y-1、杂质Y-2、杂质Y-3、杂质Y-4、杂质M1各约5 mg和杂质A、杂质D、杂质F、杂质G、杂质K、杂质N氧化物各约1 mg,精密称定,分别置10 mL量瓶中,用甲醇溶解并稀释至刻度,摇匀,即得各杂质溶液。取甲苯磺酸索拉非尼原料约15 mg,精密称定,精密量取各杂质溶液0.5 mL,置同一10 mL量瓶中,用稀释剂溶解并稀释至刻度,摇匀,即得混合溶液。按上述色谱条件测定。图谱见图 2,该色谱条件下索拉非尼峰与相邻杂质峰,相邻杂质峰与杂质峰之间分离度良好。

|
图 2 分离度图谱 Figure 2 Chromatogram of resolution |
a)未破坏:取甲苯磺酸索拉非尼原料约37.5 mg,精密称定,置25 mL量瓶中,用稀释剂溶解并稀释至刻度,摇匀,按上述色谱条件测定。b)酸破坏:取甲苯磺酸索拉非尼原料药约37.5 mg,精密称定,置25 mL量瓶中,精密加入稀释剂10 mL,振摇1 min,加入1 mol·L-1盐酸溶液2 mL,室温放置72 h,用1 mol·L-1氢氧化钠溶液中和溶液至pH 5~7,超声2 min,用稀释剂稀释至刻度,摇匀,按上述色谱条件测定。c)过氧化氢氧化破坏:取甲苯磺酸索拉非尼原料药约37.5 mg,精密称定,置25 mL量瓶中,精密加入稀释剂10 mL,振摇1 min,加入30%过氧化氢溶液4 mL,室温放置72 h,密封,取出,超声2 min,用稀释剂稀释至刻度,摇匀,按上述色谱条件测定。d)高温破坏:取甲苯磺酸索拉非尼原料药约37.5 mg,精密称定,置25 mL量瓶中,加稀释剂约20 mL,放置于60 ℃电热干燥箱中,放置5 d,取出,用稀释剂稀释至刻度,摇匀,按上述色谱条件测定。e)光照破坏:取甲苯磺酸索拉非尼原料药约37.5 mg,精密称定,置25 mL量瓶中,加稀释剂约20 mL,放置于光照下照射,总照度不小于1.2×106lx(根据ICH指导原则),取出,用稀释剂稀释至刻度,摇匀,按上述色谱条件测定。f)碱破坏:取甲苯磺酸索拉非尼原料药约37.5 mg,精密称定,置25 mL量瓶中,精密加入稀释剂10 mL,振摇1 min,加入1 mol·L-1氢氧化钠溶液2 mL,室温放置72 h,用1 mol·L-1盐酸溶液中和溶液至pH 5~7,超声处理(功率300 W、频率40 kHz)2 min,用稀释剂稀释至刻度,摇匀,按上述色谱条件测定。
上述破坏试验色谱图见图 3,结果表明,本品原料在酸、碱、氧化、光照条件均较稳定,在高温条件下产生明显的降解产物,其主要降解产物为杂质A、D、F、M1,所产生的杂质峰与主成分峰均能达到有效分离,表明方法的专属性强。

|
a.未破坏(undestroyed)b.酸破坏(destroyed by acid)c.氧化破坏(destroyed by oxidation)d.高温破坏(destroyed by high temperature)e.光照破坏(destroyed by light)f.碱破坏(destroyed by alkaline) 图 3 专属性试验图谱 Figure 3 Chromatograms of specificity tests |
杂质A、D、F、M1的质量浓度分别为0.06、0.17、0.14、0.06 μg·mL-1的溶液信噪比约为10,作为定量下限浓度;浓度分别为0.01、0.05、0.03、0.01 μg·mL-1的溶液信噪比约为3,作为检测下限浓度。
2.4.3 线性与范围杂质A溶液配制:精密称定10 mg置10 mL量瓶,用稀释剂稀释定容至刻度,精密量取上述溶液3 mL置25 mL量瓶,用稀释剂稀释定容至刻度,再取该溶液9 mL置200 mL量瓶,用稀释剂稀释定容至刻度,即得400%母液。杂质D、F、M1溶液配制同杂质A溶液。
甲苯磺酸索拉非尼溶液配制:精密称定15 mg置10 mL量瓶,用稀释剂溶解并定容至刻度,精密量取上述溶液1 mL置25 mL量瓶,用稀释剂定容至刻度,再取该溶液5 mL置50 mL量瓶,用稀释剂定容至刻度,即得400%的母液。
线性溶液的配制:分别精密量取母液2、3、4、5、10 mL,置20 mL量瓶中,用稀释剂定容至刻度,摇匀。分别精密量取各稀释液5 μL,注入液相色谱仪,记录色谱图。以浓度为横坐标、峰面积为纵坐标进行线性回归。各线性与范围结果如表 3。
|
|
表 3 线性与范围测定结果 Table 3 The results of linear and range |
杂质A、D、F、M1质量浓度分别在0.567 0~5.670 2、0.569 1~5.691 0、0.573 5~5.734 8、0.556 4~5.564 4μg·mL-1范围内线性关系良好,校正因子分别为3.2、1.1、1.6、2.1,测定各杂质含量时分别除以各自的校正因子[17-18]。
2.4.4 精密度甲乙两人分别照“2.2.1”项下方法配制6份供试品溶液,在不同时间,采用仪器系统1与仪器系统2分别进样,测得索拉非尼含量的平均值(n=6)分别为93.01%(RSD=0.4%)、93.05%(RSD=0.6%),12份供试品的杂质峰个数和质量基本一致,表明该方法精密度良好。
2.4.5 回收率分别取甲苯磺酸索拉非尼工作对照品约30 mg,精密称定,置20 mL量瓶中,精密量取“2.2.2”项下杂质溶液4、5、6 mL置量瓶中,用稀释剂溶解并稀释至刻度,摇匀,配制成含杂质80%、100%、120%的混合溶液各3份。取上述溶液各5 μL进样,记录色谱图,计算回收率,杂质A的平均回收率为99.5%,RSD为0.69%;杂质D的平均回收率为96.7%,RSD为0.98%;杂质F的平均回收率为99.3%,RSD为0.65%;杂质M1的平均回收率为93.3%,RSD为1.9%。
2.4.6 溶液稳定性取甲苯磺酸索拉非尼适量,按供试品溶液的制备方法操作,制成的供试品溶液于室温条件下放置,分别在0、2、4、6、8、10、12、24、36、48 h时各进样5 μL。结果杂质质量和个数均未增加,表明供试品溶液在48 h内稳定。
2.4.7 耐用性通过改变柱温(33/37 ℃)、流速(0.9/1.1 mL·min-1)和初始流动相比例(82:18)/(78:22),照有关物质测定方法进行测定。结果表明,色谱条件作微小变动,其分离效果、检出的杂质峰个数未发生明显变化,该方法耐用性良好。
2.5 样品有关物质的测定取3批自制原料适量,制备供试品溶液,进样分析,记录色谱图。3批样品的有关物质测定结果见表 4。
|
|
表 4 有关物质测定结果(%) Table 4 The result of determination of related substances |
在专利N201510578004.2的一种采用高效液相色谱法测定甲苯磺酸索拉非尼含量和有关物质的方法[19]中使用0.1%至1%的三氟乙酸作为流动相A,较大浓度的三氟乙酸对色谱柱的伤害较大,且三氟乙酸价格昂贵。甲磺酸索拉非尼片进口注册标准[20]含量测定项下的色谱条件亦可检测有关物质,但流动相A为纯缓冲盐溶液,容易长菌。本研究流动相采用一定比例的磷酸盐缓冲液-乙腈-乙醇溶液,对色谱柱没有伤害,延长了色谱柱使用寿命,且磷酸二氢钾价格低,检测成本低,且不易长菌,普适性好。
3.2 杂质归属与降解途径由强制降解试验可知,甲磺酸索拉非尼在酸、碱、氧化、光照条件下较稳定,在高温条件下产生明显的降解产物。
通过外购得到杂质A、D、F,自制得到杂质M1对照品,并对其结构进行了确证,在相同色谱条件下,通过保留时间确认甲磺酸索拉非尼在高温条件下产生主要降解产物杂质A、D、F、M1,分别为乙醇解产物、水解产物、乙醇解产物、过程中间体,与甲磺酸索拉非尼片进口注册标准中提到的相应降解产物出峰顺序一致,故确定杂质A、D、F、M1,为高温降解杂质,需在长期稳定性试验过程中着重监控。
3.3 杂质控制[21]由于杂质A、D、F、M1较难获得,故对其采用相对保留时间定位,计算采用加校正因子的主成分自身对照法进行计算;经试验,测定其相对保留时间分别为0.53、0.71、0.96、0.14,校正因子分别为3.2、1.1、1.6、2.1。
综上所述,本试验中采用高效液相色谱法测定自制甲苯磺酸索拉非尼中有关物质,经方法学研究显示,该方法专属、准确且普适性好,可用于本品有关物质测定,为该药的进一步研究开发提供了有效的质量控制方法。
| [1] |
王玉兰, 林挺岩, 陈湘琦. 索拉非尼抗肿瘤作用的研究现状与进展[J]. 中国医药指南, 2011, 33(9): 273. WANG YL, LIN TY, CHEN XQ. Research status and progress of antitumor effect of sorafenib[J]. Guide China Med, 2011, 33(9): 273. DOI:10.3969/j.issn.1671-8194.2011.09.208 |
| [2] |
陆志城. 索拉非尼: 首个获准上市的多靶点靶向治疗药物[N]. 医药经济报, 2006-07-07(A06) LU ZC. Sorafenib: The first targeted multi-targeted therapeutic drug[N]. Medical Economic News, 2006-07-07(A06) |
| [3] |
徐述湘. 索拉非尼抗肾细胞癌获FDA批准[N]. 中国医药报, 2006-04-04(A05) XU SX. Sorafenib anti-renal cell carcinoma was approved by the FDA[N]. China Pharmaceutical News, 2006-04-04(A05) |
| [4] |
拜耳公司抗肝癌药物索拉非尼获准进入中国[J]. 世界临床药物, 2008, 29(9): 537 Bayer's anti-hepatoma drug sorafenib being approved to enter China[J]. World Clin Drugs, 2008, 29(9): 537 http://lib.cqvip.com/qk/81668X/200001/29747703.html |
| [5] |
唐克, 李燕, 陈晓光. 多靶点抗肿瘤药物索拉非尼的研究进展[J]. 中国新药杂志, 2011, 20(24): 2434. TANG K, LI Y, CHEN XG. Advance in the research of sorafenib, a multiple targeted antitumor agent[J]. Chin J New Drugs, 2011, 20(24): 2434. |
| [6] |
郑玮, 寿建忠, 马建辉. 索拉非尼治疗肾癌的研究现状[J]. 临床药物治疗杂志, 2012, 10(3): 19. ZHENG W, SHOU JZ, MA JH. Research status of sorafenib for patients with renal cell carcinoma[J]. Clin Med J, 2012, 10(3): 19. DOI:10.3969/j.issn.1672-3384.2012.03.005 |
| [7] |
燕强勇, 赵云丽, 闫晗, 等. HPLC加校正因子的主成分自身对照法测定埃索美拉唑镁的有关物质[J]. 沈阳药科大学学报, 2013, 30(10): 782. YAN QY, ZHAO YL, YAN H, et al. Determination of related substances in esomeprazole magnesium by HPLC method using correction factor[J]. J Shenyang Pharm Univ, 2013, 30(10): 782. |
| [8] |
鹿贵花, 杨梅, 胡丽娜. 加校正因子的主成分自身对照法同时测定咪喹莫特5个杂质[J]. 药物分析杂志, 2017, 37(7): 1320. LU GH, YANG M, HU LN. Simultaneous determination of five impurities of imiquimod by self-contrast method with principal component of correction factor[J]. Chin J Pharm Anal, 2017, 37(7): 1320. |
| [9] |
李建伟, 宋素丽, 申爱卓. 加校正因子的主成分自身对照法测定复方片剂中瑞格列奈有关物质[J]. 药物分析杂志, 2015, 35(8): 1488. LI JW, SONG SL, SHEN AZ. Determination of repaglinide related substances in compound tablets with the correction factor and self-contrast method[J]. Chin J Pharml Anal, 2015, 35(8): 1488. |
| [10] |
朱雪萍, 陈爱萍, 戴云志. 加校正因子的主成分自身对照法测定复方依折麦布瑞舒伐他汀钙片的有关物质[J]. 中国药学杂志, 2017, 52(2): 140. ZHU XP, CHEN AP, DAI YZ. Determination of related substances of compound ezetimibe rosuvastatin calcium tablet by self-contrast method with correction factor[J]. Chin Pharm J, 2017, 52(2): 140. |
| [11] |
刘琳, 李慧, 张菁, 等. 加校正因子的主成分自身对照法测定青霉素V钾片有关物质[J]. 中国药品标准, 2017, 18(1): 29. LIU L, LI H, ZHANG J, et al. The method of main component self-control corrected with calibration factor was used for determination of related substances in phenoxymethylpenicillin potassium tablets[J]. Drug Stand China, 2017, 18(1): 29. |
| [12] |
丁锐, 纪宏, 陈思, 等. 用加校正因子的主成分自身对照法测定注射用前列地尔中有关物质的含量[J]. 中国药科大学学报, 2010, 41(5): 462. DING R, JI H, CHEN S, et al. Assay of the related substances in alprostadil for injection with the correction factor[J]. J China Pharm Univ, 2010, 41(5): 462. |
| [13] |
魏嘉陵, 林畅伟. 用加校正因子的主成分自身对照法测定苯扎贝特中杂质氯苯酪胺的含量[J]. 药物分析杂志, 2006, 26(12): 1848. WEI JL, LIN CW. The determination of the content of impurity chlorobenzoyltyramine in bezafibrate with the correction factor for the substance and impurities being examined[J]. Chin J Pharm Anal, 2006, 26(12): 1848. |
| [14] |
邓晶晶, 张春然, 唐克慧, 等. 反相高效液相色谱法测定头孢美唑钠中的有关物质[J]. 中国新药杂志, 2011, 20(9): 828. DENG JJ, ZHANG CR, TANG KH, et al. Determination of the related substances in cefmetazole sodium by RP-HPLC[J]. Chin J New Drugs, 2011, 20(9): 828. |
| [15] |
齐敬敬, 孙春艳, 褚岩凤, 等. HPLC法测定吉非替尼原料药有关物质[J]. 药物分析杂志, 2016, 36(4): 704. QI JJ, SUN CY, CHU YF, et al. Determination of gefitinib related substances by HPLC[J]. Chin J Pharm Anals, 2016, 36(4): 704. |
| [16] |
中华人民共和国药典2015年版. 四部[S]. 2015: 374 ChP 2015. Vol Ⅳ[S]. 2015: 374 |
| [17] |
左文飞, 潘娜, 范雪平, 等. 泮托拉唑钠杂质校正因子的测定[J]. 中国药学杂志, 2012, 47(24): 2029. ZUO WF, PAN N, FAN XP, et al. Determination of impurities in pantoprazole sodium[J]. Chin Pharm J, 2012, 47(24): 2029. |
| [18] |
朱静毅, 李静, 闻琍毓, 等. 校正因子法同时测定非洛地平及其片剂中的3个杂质[J]. 药物分析杂志, 2014, 34(2): 281. ZHU JY, LI J, WEN LM, et al. Simultaneous quantification of three impurities in felodipine and its tablets by HPLC method with correction factor[J]. Chin J Pharm Anal, 2014, 34(2): 281. |
| [19] |
黄乐群, 曹梅. 一种高效液相色谱法测定甲苯磺酸索拉非尼含量和有关物质的方法: 中国, CN105181844A[P]. 2015-12-23 HUANG LQ, CAO M. A Method for the Determination of Sorafenib Tosylate and Related Substances by High Performance Liquid Chromatography: China, CN105181844A[P]. 2015-12-23 |
| [20] |
JX20070240国家食品药品监督管理局进口药品注册标准[S]. 2008 JX20070240 State Food and Drug Administration Import Drug Registration Standard[S]. 2008 |
| [21] |
张玲, 李纬, 陆小平. 关于HPLC法测定药品中杂质含量的讨论[J]. 中国药品标准, 2002, 3(6): 10. ZHANG L, LI W, LU XP. Discussion on HPLC method of determining impurities in medicines[J]. Drug Stand China, 2002, 3(6): 10. DOI:10.3969/j.issn.1009-3656.2002.06.004 |