 2018, Vol. 38
2018, Vol. 38 

2. 江西省药品与医疗器械质量工程技术研究中心, 南昌 330029
2. Jiangxi Provincial Engineering Research Center for Drug and Medical Device Quality, Nanchang 330029, China
棓丙酯是中药赤芍的主要成分没食子酸经酯化反应合成的具有更强生物效应的单体药物,具有抑制血栓素A2引起的血小板聚集,降低血液黏度和扩张血管平滑肌的作用,临床用于预防和治疗心脑血管疾病[1]。棓丙酯注射剂包括注射用棓丙酯、棓丙酯注射液和棓丙酯氯化钠注射液3种剂型。注射用棓丙酯现行质量标准为中国药典2015年版二部[2]和国家食品药品监督管理总局国家药品标准WS-10001-(HD-0109)-2002-2014;棓丙酯注射液质量标准收载于国家食品药品监督管理总局国家药品标准WS1-XG-008-2014;棓丙酯氯化钠注射液质量标准收载于国家食品药品监督管理总局国家药品标准WS1-XG-013-2014。3个剂型4个标准中均设置了有关物质检查项,且各标准检查方法一致,均为采用HPLC法测定没食子酸及其他未知杂质的量。国外药典未收载棓丙酯有关物质的HPLC检测法。多篇文献采用HPLC法测定了棓丙酯注射液中有关物质和已知杂质没食子酸[3-7],但均未对其他杂质进行定性研究。
目前对未知杂质进行定性研究的方法较多,主要为质谱、核磁、红外、紫外、熔点等,其中通过分离纯化制备得到杂质单体后再进行四大光谱分析,确证出的结构更为准确,但制备含量较小的杂质较为困难,而通过色谱-质谱联用技术进行结构推断则不需要制备杂质纯品,更加简单、快速。目前已有多篇文献[8-10]报道了色谱-质谱联用技术应用于有关物质的结构推断。棓丙酯分子结构中含有酯键,易水解成没食子酸[7],且其具有连苯三酚结构,容易发生自氧化[11]和亲电取代反应[12],容易产生杂质。本研究采用LC-DAD-MS/MS法对22家企业生产棓丙酯注射剂(注射用棓丙酯、棓丙酯注射液和棓丙酯氯化钠注射液)中的有关物质进行了测定,结果6家企业的注射用棓丙酯均检出同一杂质,为方便表述,将其命名为杂质Ⅰ。此外,1批棓丙酯氯化钠注射液检出另1个较大的杂质,将其命名为杂质Ⅱ。提取杂质Ⅰ、Ⅱ的紫外光谱图、母离子扫描图和子离子扫描图,进行结构推测,并进一步采用LC-TOF-MS扫描精确质量数,检索匹配的分子式,综合上述信息,并结合处方工艺以及国内外参考文献,对杂质Ⅰ、Ⅱ的结构进行了推测。通过模拟实验研究和处方工艺的对比分析,推测了杂质Ⅰ、Ⅱ产生的来源。
1 仪器与试剂Waters Xevo TQ-S型液相色谱-串联四极杆质谱仪(沃特世公司):Waters Acquity UPLC自动进样器;Waters Acquity UPLC四元泵;Waters Acquity UPLC柱温箱;Waters Acquity UPLC PDA二极管阵列检测器;软件MassLynx V 4.1版;ACQUITY UPLC BEH C18色谱柱(填料:十八烷基硅烷键合硅胶;2.1 mm×50 mm,1.7 μm;沃特世公司);Waters Xevo G2Q-TOF型飞行时间质谱仪(沃特世公司):Waters Acquity UPLC自动进样器;Waters Acquity UPLC四元泵;Waters Acquity UPLC柱温箱;软件MassLynx V 4.1版。
甲酸、乙腈、甲酸均为色谱纯,水为纯化水;棓丙酯对照品由中国食品药品检定研究院提供,批号100407-200301;注射用棓丙酯(13批)分别来自于A~M共13家生产企业,A企业产品规格为120 mg,B~M企业产品规格为60 mg;棓丙酯氯化钠注射液(3批)分别来自于N~P 3家生产企业,规格为250 mL:棓丙酯0.18 g与氯化钠2.25 g。棓丙酯注射液(6批)来自于为Q~V 6家生产企业,Q和R企业产品规格为5 mL:180 mg,S企业产品规格为2 mL:60 mg,T企业产品规格为5 mL:0.12 g,U企业产品规格为5 mL:60 mg,V企业产品规格为10 mL:0.18 g。棓丙酯原料由A企业提供。
2 方法 2.1 色谱条件应用ACQUITY UPLC BEH C18色谱柱(2.1 mm×50 mm,1.7 μm),柱温25 ℃,以乙腈-0.05%甲酸(20 :80)为流动相,流速0.2 mL·min-1,PDA检测波长为272 nm,扫描波长为190~400 nm,进样量2 μL。
2.2 质谱条件电喷雾负离子化(ESI-)检测,去溶剂温度500 ℃,干燥气流量10 L·h-1,毛细管电压1.5 kV,锥孔电压40 V,质量数扫描范围m/z 100~800,碰撞电压40 V。
2.3 对照品溶液的制备取棓丙酯对照品10.12 mg,置100 mL量瓶中,加甲醇溶解并稀释至刻度,摇匀,精密量取5 mL,置50 mL量瓶中,用甲醇稀释至刻度,摇匀,即得10.12 μg·mL-1的棓丙酯对照品溶液。
2.4 供试品溶液的制备棓丙酯原料:取样品,用甲醇溶解并稀释制成每1 mL中约含棓丙酯0.6 mg的溶液,经0.22 μm滤膜过滤,即得。
注射用棓丙酯:取样品,用甲醇溶解并稀释制成每1 mL中约含棓丙酯0.6 mg的溶液,经0.22 μm滤膜过滤,即得。
棓丙酯氯化钠注射液:取样品,经0.22 μm滤膜过滤,即得。
棓丙酯注射液:取样品适量,用甲醇定量稀释并制成每1 mL中约含棓丙酯0.6 mg的溶液,经0.22 μm滤膜过滤,即得。
3 结果 3.1 色谱图与测定结果 3.1.1 LC-PDA-ESI-MS/MS取棓丙酯原料、注射用棓丙酯、棓丙酯注射液和棓丙酯氯化钠注射液的供试品溶液,按“2.1”和“2.2”项下条件,进行LC-PDA-ESI-MS/MS分析,为避免污染离子源,将0 ~0.9 min和3.6~4.2 min通过阀切换到废液,结果A~F共6家企业的注射用棓丙酯均在4.4 min左右检出同一杂质,该杂质在棓丙酯注射液、棓丙酯氯化钠注射液和棓丙酯原料中均未检出。为方便表述,将其命名为杂质Ⅰ。此外,1批N企业生产的棓丙酯氯化钠注射液在2.4 min检出另1个较大的杂质,将其命名为杂质Ⅱ,该杂质在其余样品及原料中均未检出,22家企业的棓丙酯注射剂和棓丙酯原料中检出杂质Ⅰ和杂质Ⅱ的情况见表 1。棓丙酯原料、粉针、注射液和氯化钠注射液的典型LC-PDA-MS/MS色谱图见图 1,提取杂质Ⅰ、Ⅱ和棓丙酯的紫外光谱图,结果见图 2。提取杂质Ⅰ和杂质Ⅱ的质谱全扫描图,结果杂质Ⅰ的母离子m/z为421,杂质Ⅱ的母离子m/z为291,分别对m/z 421和m/z 291进行子离子扫描,结果见图 3。
|
|
表 1 棓丙酯原料和制剂中杂质Ⅰ和杂质Ⅱ的检测结果 Table 1 The test results of impurities Ⅰ and Ⅱ in propylgallate and propylgallate preparation |

|
A. A企业的注射用棓丙酯(propylgallate for injection coming from company A)B. N企业的棓丙酯氯化钠注射液(propylgallate sodium chloride injection coming from company N)C. Q企业的棓丙酯注射液(propylgallate injection coming from company Q)D.棓丙酯(propylgallate coming from company A) 图 1 棓丙酯原料及制剂的典型色谱图 Figure 1 Typical chromatograms of propylgallate and propylgallate preparation |

|
图 2 杂质Ⅰ、Ⅱ和棓丙酯的紫外光谱图 Figure 2 Ulnivitor spectrums of the(impurity)Ⅰ、Ⅱ and propylgallate |

|
图 3 杂质Ⅰ和杂质Ⅱ的子离子扫描图 Figure 3 Product MS spectra of related substances Ⅰ and Ⅱ |
取10.12 μg·mL-1的棓丙酯对照品溶液,按“2.1”和“2.2”项下条件进行LC-TOF-MS分析,结果棓丙酯出峰时间为4.89 min,其准分子离子为[M-H]-峰,m/z为211.059 8。取含杂质Ⅰ的A企业注射用棓丙酯样品1批,用甲醇配制成约含棓丙酯0.6 mg·mL-1的溶液,按“2.1”和“2.2”项下条件进行LC-TOF-MS分析,因棓丙酯浓度过高,且样品中含不挥发性盐等污染物,为避免污染离子源,将0~1.8 min和4.4~5.4 min通过阀切换到废液,结果杂质Ⅰ出峰时间为5.95 min,其负离子全扫描m/z为421.113 1。取含杂质Ⅱ的N企业棓丙酯氯化钠注射液样品1批,按“2.1”和“2.2”项下条件进行LC-TOF-MS分析,结果杂质Ⅱ出峰时间为2.46 min,其负离子全扫描丰度最大的m/z为291.016 9,其次为m/z 211.059 8和312.997 7。棓丙酯原料及制剂的LC-TOF-MS总离子流图见图 4,质谱图见图 5,对杂质Ⅰ和杂质Ⅱ的准分子离子进行分子式解析,结果见表 2。

|
A.棓丙酯(propylgallate coming from company A)B. A企业的注射用棓丙酯(propylgallate for injection coming from company A)C. N企业的棓丙酯氯化钠注射液(propylgallate sodium chloride injection coming from company N) 图 4 棓丙酯及其制剂LC-TOF-MS总离子流图 Figure 4 TIC of propylgallate and propylgallate preparation |

|
图 5 棓丙酯及杂质Ⅰ、Ⅱ质谱图 Figure 5 MS spectra of propylgallate and rmpuriiesⅠand Ⅱ |
|
|
表 2 LC-TOF-MS测定结果 Table 2 Test results of LC-TOF-MS |
根据杂质Ⅰ、Ⅱ的LC-PDA-ESI-MS/MS和LC-TOF-MS测定结果,综合其紫外光谱图、解析的准分子离子的分子式、子离子碎片信息以及制剂处方工艺,推测杂质Ⅰ为棓丙酯二聚物,杂质Ⅱ为棓丙酯磺酸化产物,推测的杂质Ⅰ和杂质Ⅱ的结构见表 3。
|
|
表 3 推测的杂质Ⅰ和杂质Ⅱ的结构 Table 3 The elucidated structure of impurity Ⅰ and Ⅱ |
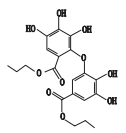
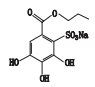
为更好地推测棓丙酯杂质的结构,首先对棓丙酯的质谱裂解途径进行推测。取10.12 μg·mL-1的棓丙酯对照品溶液,按“2.1”及“2.2”项下条件进行分析,对m/z 211进行子离子扫描,结果见图 6。从图可知棓丙酯负离子条件下产生的碎片离子为m/z 169、m/z 151、m/z 125和m/z 124,与文献[13]报道的相符,据文献报道,棓丙酯具有连苯三酚的结构,易发生自氧化,首先由负离子形式(-O-R-O-)与氧气作用发生电子转移形成氧自由基(·O-R-O-和

|
图 6 棓丙酯子离子扫描结果 Figure 6 The product MS spectrum of the propylgallate |

|
图 7 棓丙酯质谱碎裂途径 Figure 7 The MS fragmentation pathway of the propylgallate |
杂质Ⅰ在C18柱上出峰时间比棓丙酯略晚,可见其极性比棓丙酯弱,杂质Ⅰ紫外图谱与棓丙酯相似,说明共轭体系未发生明显变化,且其质谱裂解碎片也与棓丙酯裂解碎片相似,根据棓丙酯结构特性、TOF-MS检索出来的分子式及杂质Ⅰ的质谱裂解碎片,推测杂质Ⅰ的结构为二分子棓丙酯聚合形成的二聚物。棓丙酯为连苯三酚类结构,连苯三酚较易形成半醌自由基,苯环上未取代的部位剩余成键能力较大,较易受到新生氧的攻击[14],因而有可能进一步反应生成偶联的二聚物。已有文献报道,邻苯酚氧化形成邻醌后可与未反应的邻苯酚反应生成邻酚类偶联产物,且邻位的酚羟基更活泼[18],因此推测杂质Ⅰ为一分子棓丙酯氧化成邻醌后由邻位的醌进攻另一分子未被氧化的棓丙酯生成的偶联产物,推测的质谱裂解途径见图 8。若一分子棓丙酯由中间的酚羟基氧化为醌后偶联另一分子的棓丙酯,则无法形成m/z 301的碎片,只能形成m/z 303的碎片离子,与实际测得结果不符,因此杂质Ⅰ只有一种可能的结构。

|
图 8 推测的杂质Ⅰ质谱裂解途径 Figure 8 The MS fragmentation pathway of impurity Ⅰ |
仅1家生产企业的棓丙酯氯化钠注射液检出杂质Ⅱ,分析该企业处方,发现其添加了亚硫酸氢钠作为辅料。根据棓丙酯的结构,在芳环的酚羟基邻位上极易发生亲电取代反应,结合杂质Ⅱ的质谱特征,推测杂质Ⅱ为棓丙酯与亚硫酸氢钠反应产生的磺酸化产物,分子式为C10H11NaO8S,相对分子质量为314.0。LC-TOF-MS结果显示,杂质Ⅱ准分子离子峰为m/z 291.016 9,LC-TOF-MS解析出的第1个结构式C10H11O8S即与推测的杂质Ⅱ(棓丙酯磺酸钠)[M-Na]-结构式一致。而杂质Ⅱ负离子全扫描中还有1个峰为m/z 312.997 7,其解析的分子式C10H11NaO8S与推测的杂质Ⅱ(棓丙酯磺酸钠)[M-H]-结构式一致,而杂质Ⅱ负离子全扫描中还有1个峰为m/z 211.059 8,与棓丙酯[M-H]-的质荷比完全一致,为杂质Ⅱ[M-NaHSO3]-。之所以出现多个质谱峰是因为杂质Ⅱ负离子条件下最容易形成[M-Na]-,另外也存在少量的[M-H]-和[M-NaHSO3]-。
由于杂质Ⅱ在棓丙酯苯环上连接了1个磺酸基,磺酸基吸电子导致苯环共轭系统的共轭程度减弱,因此波长蓝移,使最大吸收波长比棓丙酯小,与其紫外光谱特征吻合。文献报道,磺酸化合物在质谱中易形成[M-H-SO3]-,同时出现明显的m/z 80的[SO3·-]碎片[19-20],根据LC-PDA-ESI-MS/MS测定结果,杂质Ⅱ母离子m/z 291,比棓丙酯母离子质荷比大了80,根据子离子扫描结果,图谱中明显可见m/z 80的碎片峰,其丰度最强的碎片m/z 211与棓丙酯母离子质荷比一致,而碎片m/z 169、m/z 124则与棓丙酯的主要碎片离子一致。推测的质谱裂解途径见图 9。这些结果进一步佐证了杂质Ⅱ结构推测的正确性。

|
图 9 推测的杂质Ⅱ质谱裂解途径 Figure 9 The MS fragmentation pathway of related substance Ⅱ |
根据推测的杂质Ⅰ结构分析,杂质Ⅰ为一分子棓丙酯氧化后再与另一分子未氧化的棓丙酯结合产生。从原料和制剂测定结果可知,杂质Ⅰ仅来自于部分企业生产的注射用棓丙酯样品,原料和注射液中均未检出,因此推测是注射用棓丙酯生产或储存过程中产生。对比所有企业注射用棓丙酯的处方工艺,发现检出杂质Ⅰ的样品企业工艺中均采用了加热使棓丙酯溶解(棓丙酯在冷水中溶解度较差),且存在保温过程,而未检出杂质Ⅰ的部分样品企业处方中添加了助溶剂,因此工艺中未加热溶解棓丙酯,其余未添加助溶剂的样品工艺中加热溶解棓丙酯后即放冷,无保温过程。再分析注射液的处方工艺,发现5家企业处方中添加了抗氧剂和助溶剂,这5家企业中部分企业工艺中有加热过程,部分企业工艺中无加热过程,1家企业处方中未添加抗氧剂,但添加了助溶剂,工艺中无加热过程。综合上述分析,推测杂质Ⅰ为棓丙酯加热过程中产生,而有抗氧剂存在下则能避免该杂质产生。根据杂质Ⅱ结构推测结果,杂质Ⅱ为棓丙酯与亚硫酸氢钠反应的产物。仅1家生产企业的棓丙酯氯化钠注射液检出杂质Ⅱ,该企业处方中添加了亚硫酸氢钠作为辅料。
为了进一步确定杂质Ⅰ和杂质Ⅱ产生的来源,分别称取5份10 mg棓丙酯原料,第1份用10 mL水加热溶解后室温放置10 d,第2份用10 mL水加热溶解后置水浴中加热4 h,第3份用10mL水加热溶解后加10 mg亚硫酸氢钠,置水浴加热4 h,第4份用10 mL水加热溶解后加10 mg亚硫酸氢钠,于室温放置4 h,第5份用10 mL水加热溶解后加10 mg亚硫酸氢钠,于室温放置10 d。将5份溶液按“2.1和2.2”项下色谱和质谱条件进行LC-DAD-MS/MS分析,结果杂质Ⅰ和杂质Ⅱ检出情况见表 4。从表中可知,棓丙酯水溶液水浴加热4 h后产生了杂质Ⅰ,而添加亚硫酸氢钠后则不会产生杂质Ⅰ。棓丙酯与亚硫酸氢钠混合4 h内未产生杂质Ⅱ,室温放置10 d后则产生了杂质Ⅱ。根据以上结果推测,杂质Ⅰ为棓丙酯在水溶液中较长时间加热后产生,而溶液中若添加了抗氧剂亚硫酸氢钠则不会产生杂质Ⅰ。杂质Ⅱ为棓丙酯与亚硫酸氢钠缓慢反应产生,应该为注射液储藏过程中产生的杂质。
|
|
表 4 棓丙酯经不同条件处置后杂质Ⅰ和杂质Ⅱ的检测结果 Table 4 The test results of related substance Ⅰ and Ⅱ in propylgallate at difference conditions |
对各制剂处方进行分析发现,棓丙酯注射液及棓丙酯氯化钠注射液处方中均添加了亚硫酸氢钠,但仅1家企业的棓丙酯氯化钠注射液检出杂质Ⅱ,该制剂在灌装工艺中未充氮气进行保护,其他制剂在灌装工艺中充了氮气,结果均未检出该杂质,推测原因为在无氧条件下棓丙酯与亚硫酸氢钠则不容易发生磺酸化反应,提示充氮可避免杂质Ⅱ的产生。
此外,所有的棓丙酯注射液工艺均采用了充氮气后高温灭菌(包括未添加抗氧剂的样品),而粉针剂均为过滤除菌,注射液中均未检出杂质Ⅰ,因此推测杂质Ⅰ须在有氧条件下加热才会产生,充氮气后加热也不会产生该杂质。
5 小结本试验建立了棓丙酯注射剂中有关物质的LC-PDA-MS/MS和LC-TOF-MS测定法,根据紫外光谱图、质谱信息,以及棓丙酯注射剂的生产工艺,推断出2个较大杂质的结构与生成途径。色谱-质谱联用技术能有效地对棓丙酯中有关物质进行结构解析,为其工艺和质量控制提供参考依据。
| [1] |
汪小荣. 棓丙酯治疗冠心病心绞痛疗效观察[J]. 实用心脑肺血管病杂志, 2006, 14(12): 978. WANG XR. Clinical efficacy of adjuvant therapy with propyl gallate in Angina Pectoris[J]. Pract J Cardiac Cereb Pneum Vasc Dis, 2006, 14(12): 978. DOI:10.3969/j.issn.1008-5971.2006.12.023 |
| [2] |
中国药典2015年版. 二部[S]. 2015: 1280 ChP 2015.Vol Ⅱ[S].2015:1280 |
| [3] |
秦向阳, 赵英永, 姜茹, 等. RP-HPLC法测定注射用棓丙酯的含量及有关物质[J]. 药物分析杂志, 2011, 31(7): 1368. QIN XY, ZHAO YY, JIANG R, et al. RP-HPLC determination of propylgallate for injection and its related substances[J]. Chin J Pharm Anal, 2011, 31(7): 1368. |
| [4] |
谭会洁, 张冬梅. HPLC法测定注射用棓丙酯中的有关物质[J]. 中国药品标准, 2010, 11(6): 456. TAN HJ, ZHANG DM. Determination of related substances in propylgallate for injection by HPLC[J]. Drug Stand China, 2010, 11(6): 456. |
| [5] |
杨本霞, 袁利杰, 李婷婷. 棓丙酯注射液有关物质检查中没食子酸测定方法的改进[J]. 中国药品标准, 2015, 16(5): 362. YANG BX, YUAN LJ, LI TT. Improvement of gallic acid determination in related substances inspection for propylgallate injection[J]. Drug Stand China, 2015, 16(5): 362. |
| [6] |
陈洪. 棓丙酯注射液中没食子酸的HPLC法测定[J]. 中国医药工业杂志, 2015, 46(3): 287. CHEN H. Determination of gallic acid in propyl gallate injection by HPLC[J]. Chin J Pharm, 2015, 46(3): 287. |
| [7] |
徐幸民, 李健和, 伍晓群, 等. 高效液相色谱法测定棓丙酯注射液含量及有关物质[J]. 中国医药导报, 2009, 6(18): 63. XU XM, LI JH, WU XQ, et al. Content determination of propylgallate injection and its related substances by HPLC[J]. Chin Med Her, 2009, 6(18): 63. DOI:10.3969/j.issn.1673-7210.2009.18.032 |
| [8] |
杨倩, 王志英, 唐素芳. 二维超高效液相色谱-QTof质谱联用技术在盐酸博来霉素杂质谱研究中的应用[J]. 药物分析杂志, 2016, 36(7): 1231. YANG Q, WANG ZY, TANG SF. Application of two-dimensional UPLC-QTof MS technology in the study of the impurity profile of bleomycin hydrochloride[J]. Chin J Pharm Anal, 2016, 36(7): 1231. |
| [9] |
朱培曦, 陈娟娟, 李会林, 等. 液质联用快速确定尼群地平原料药中的杂质[J]. 药物分析杂志, 2012, 32(1): 88. ZHU PX, CHEN JJ, LI HL, et al. Characterization of impurities in nitrendipine bulk drug by liquid chromatography/ion trap mass spectrometry[J]. Chin J Pharm Anal, 2012, 32(1): 88. |
| [10] |
张玉峰, 史培颖, 程翼宇. 醋酸氟孕酮原料药相关物质分析[J]. 药物分析杂志, 2007, 27(11): 1735. ZHANG YF, SHI PY, CHENG YY. Study on the related substances of flugestrone acetate[J]. Chin J Pharm Anal, 2007, 27(11): 1735. |
| [11] |
RAMASARMA T, RAO AVS, DEVI MM, et al. New insights of superoxide dismutase inhibition of pyrogallol autoxidation[J]. Mol Cell Biochem, 2015, 400(11): 277. |
| [12] |
王积涛, 张宝申, 王永梅, 等. 有机化学[M]. 第2版. 天津: 南开大学出版社, 2003, 298. WANG JT, ZHANG BS, WANG YM, et al. Organic Chemistry[M]. 2nd Ed. Tianjing: Nankai University Press, 2003, 298. |
| [13] |
GAO SH, ZHAN Q, LI JX, et al. LC-MS/MS method for the simultaneous determination of ethyl gallate and its major metabolite in rat plasma[J]. Biomed.Chromatogr, 2010, 24(8): 472. |
| [14] |
杨融生, 蔡金万, 孙燕琼, 等. 连苯三酚氧化产物及其金属配合物的合成及表征[J]. 光谱学与光谱分析, 2000, 20(5): 645. YANG RS, CAI JW, SUN YQ, et al. Synthesis and characterization of the oxidation product of the 1, 2, 3-trihydroxybenzene and its metalic complexes[J]. Spectrosc Spect Anal, 2000, 20(5): 645. |
| [15] |
王晔. 酚醌类有机小分子电化学和红外光谱电化学研究[D]. 合肥: 安徽大学, 2007 WANG Y.Electrochemistry and In-situ FTIR Spectrochemistry Studies on Paeonol and A Series of Quinones[D].Hefei:Anhui University, 2007 http://www.wanfangdata.com.cn/details/detail.do?_type=degree&id=Y1192232 |
| [16] |
夏志林. 没食子酸酯金属(Zn、Zr)配合物合成与性质研究[D]. 武汉: 武汉科技大学, 2008 XIA ZL.The Synthesis and Characterization of the Esterified Gallate-Zn and Esterified Gallate-Zr[D].Wuhan:Wuhan University of Science and Technology, 2008 http://d.wanfangdata.com.cn/Thesis_Y1322524.aspx |
| [17] |
TANG CM, TAN JH, JIN JB, et al. Observation and confirmation of oxidation reactions occurring on ultra-high-performance liquid chromatography columns[J]. Rapid Commun Mass SP, 2015, 29(20): 1863. DOI:10.1002/rcm.v29.20 |
| [18] |
裴丽霞. 邻醌型活性中间体介导的偶联环合反应及其在有机合成中的应用[D]. 广州: 中山大学, 2005 PEI LX.Active o-Quinones Mediated Coupling-Cyclization Reaction and Its Application in Organic Synthesis[D].Guangzhou:Sun Yat-Sen University, 2005 |
| [19] |
NAGESWARA RR, VENKATESWARLU N, KHALID S, et al. Use of solid-phase extraction, reverse osmosis and vacuum distillation for recovery of aromatic sulfonic acids from aquatic environment followed by their determination of using liquid chromatography-electrospray ionization tandem mass spectrometry[J]. J Chromatogr A, 2006, 1113(1): 20. |
| [20] |
FORNARINI S.Mass Spectrometry Sulfonic Acids and Their Derivatives[D].Rome, Italy:Dipartimento di Studi di Chimica e Tecnologia delle Sostanze Biologicamente Attive, Universita di Roma 'La Sapienza', 1991
|