 2017, Vol. 37
2017, Vol. 37 
咪喹莫特是一种新型免疫调节剂[1]。目前在国内外已广泛用于治疗尖锐湿疣[2]、扁平疣[3]、生殖器疱疹,各种血管瘤及病理性瘢痕[4],传染性软疣和浅表性基底细胞癌等[5],咪喹莫特收载于新药转正标准WS1-(X-125)-2005Z[6]和WS1-(X-125)-2005Z-2010[7],国外药典USP39[8]有收载。
新药转正标准WS1-(X-125)-2005Z和WS1-(X-125)-2005Z-2010标准中有关物质检查杂质限度规定比较简单,且并未详细考察具体的杂质,各杂质与主成分以及杂质之间能否有效分离未做深入的研究,USP39咪喹莫特质量标准中有关物质控制了3个已知杂质,分别为杂质A、杂质B、杂质C,2011年美国药典论坛发布的咪喹莫特质量标准征求意见稿[9]中,有关物质方法二控制了已知杂质D和杂质E,根据文献报道的咪喹莫特的合成工艺[10-12],杂质A、B、C、E为合成咪喹莫特的中间体,易带入到成品中,杂质D为合成过程可能产生的副产物,有必要对以上5个杂质分别进行定量研究,以保证药品的质量。USP39和2011年USP论坛均采用外标法对杂质进行定量检测,目前中国食品药品检定研究院没有咪喹莫特杂质对照品出售,必须购买进口的USP对照品,价格昂贵且购买周期长,造成咪喹莫特的检测费用高。
本实验对新药转正标准WS1-(X-125)-2005Z-2010咪喹莫特有关物质方法进行优化,并对优化后方法进行方法学验证,测定了咪喹莫特5个已知杂质(杂质A、B、C、D、E)的相对校正因子,首次提出加校正因子的主成分自身对照法测定本品有关物质。
1 仪器与试药Agilent 1260高效液相色谱仪(安捷伦公司);Mettler Toledo XS205分析天平(梅特勒-托利多公司)。色谱柱Waters XBridge C18(4.6 mm×250 mm,5 μm,填料:十八烷基硅烷键合硅胶,沃特世公司)。
咪喹莫特对照品(批号F0K063,USP提供,含量99.8%),咪喹莫特原料药(批号12091921、12092121、12092221,扬子江药业集团江苏海慈生物药业有限公司提供),杂质A对照品(批号F0K091,USP提供,含量100.0%),杂质B对照品(批号F0K093,USP提供,含量99.0%),杂质C对照品(批号F0K100,USP提供,含量99.0%),杂质D对照品(批号F0K106,USP提供,含量97.0%),杂质E对照品(批号F0K114,USP提供,含量99.0%)。
2 方法与结果 2.1 溶液的配制 2.1.1 混合杂质对照品溶液取咪喹莫特杂质A、B、C、D和E对照品各10 mg分别置100 mL量瓶中,加稀释剂溶解并定容至刻度,作为各杂质对照品储备液;再精密量取各杂质对照品储备液1 mL置同一100 mL量瓶中加稀释剂至刻度,摇匀,即得混合对照品溶液。
2.1.2 分离度试验溶液精密称取咪喹莫特对照品10 mg置100 mL量瓶中,加混合杂质对照品1.5 mL,加稀释剂定容至刻度,即得分离度试验溶液。
2.1.3 供试品溶液本品10 mg,精密称定,置100 mL量瓶中,加稀释剂溶解并定容至刻度,摇匀即得。临用新制。
2.1.4 对照溶液精密量取供试品溶液1 mL,置100 mL量瓶中,加稀释剂稀释至刻度,摇匀,再精密量取1 mL,置10 mL量瓶中,加稀释制成每1 mL中含咪喹莫特0.1 μg的溶液,作为对照溶液。
2.2 色谱条件及系统适用性试验采用色谱柱Waters XBridge C18(4.6 mm×250 mm,5 μm,填料:十八烷基硅烷键合硅胶);流动相:0.02 mol·L-1磷酸氢二钾缓冲液(用磷酸调节pH至8.0)-乙腈(70:30);流速:1.0 mL·min-1;柱温:25 ℃;检测波长:238 nm;进样量:10 μL;稀释剂:0.02 mol·L-1磷酸溶液-乙腈(70:30)。
精密吸取分离度试验溶液10 μL,进样。理论板数按咪喹莫特峰计算不低于10 000,咪喹莫特峰与杂质A峰的分离度应大于2.0。
2.3 检测波长选择将咪喹莫特、杂质A、B、C、D和E分别进行紫外光谱扫描[13],结果显示在238 nm波长处,各已知杂质具有较好的吸收,为了能更好地检测到各杂质,因此选择238 nm作为检测波长。
2.4 分离度试验精密量取分离度试验溶液、供试品溶液10 μL注入液相色谱仪,结果见图 1。分离度试验结果表明,各杂质之间及各杂质与主成分之间均能完全分离,分离度大于2.0,满足分离要求;理论板数按咪喹莫特峰计大于10000,方法可行。

|
1.杂质B(impurity B)2.杂质D(impurity D)3.杂质A(impurity A)4.咪喹莫特(imiquimod)5.杂质E(impurity E)6.杂质C(impurity C) A.空白溶液(blank solution)B.供试品溶液(test solution)C分离度溶液(resolution) 图 1 分离度试验色谱图 Figure 1 The chromatograms of resolution test |
将各杂质对照品储备液用稀释剂逐级稀释后进样,分别以信噪比S/N=3和10计算咪喹莫特以及杂质A、B、C、D和E的检测限和定量限,结果见表 2。
|
|
表 1 咪喹莫特及5种已知杂质结构和化学名称 Table 1 The structure and chemical name of imiquimod and five known impurities |
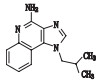
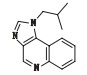
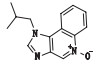
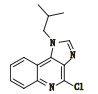
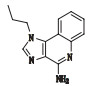
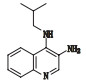
|
|
表 2 定量限和检测限结果 Table 2 Results of limit of quantification and limit of detection |
根据咪喹莫特和各杂质的定量限结果,精密称取咪喹莫特、杂质A、杂质B、杂质C、杂质D和杂质E对照品各适量,加稀释剂溶解并稀释制成浓度分别为定量限、0.06、0.09、0.12、0.15、0.18、0.24、0.3 μg·mL-1的系列溶液,分别采用2种仪器系统(1为Agilent 1260,2为Agilent 1100)进样测定,以浓度(C)为横坐标,峰面积(A)为纵坐标进行线性回归,主成分(咪喹莫特)与杂质回归方程斜率之比即为校正因子f。结果见表 3。
|
|
表 3 咪喹莫特杂质相对校正因子 Table 3 Relative correction factors of imiquimod impurities |
精密称取同一批咪喹莫特原料药(批号:12091921)9份各10 mg,置于100 mL量瓶中,加入各杂质对照品分别制成相当于供试品浓度定量限、0.15%、0.18%的混合溶液,每个浓度平行配制3份。按“2.1”项下色谱条件进样测定各杂质含量,计算回收率和RSD,结果见表 4。杂质A、B、C、D和E平均回收率分别为98.5%、97.3%、95.9%、96.2%、98.1%,均在80%~120%之间,RSD均小于5%,回收率良好。
|
|
表 4 回收率试验结果(n=9,%) Table 4 Results of the recovery test |
取本品,置光照下放置5 d。破坏结束,取光照破坏样品10.36 mg,置100 mL量瓶中,加稀释剂溶解并稀释至刻度,摇匀,作为光照破坏溶液。
2.8.2 紫外光破坏取本品,置紫外灯下放置5 d。破坏结束,取破坏样品10.17 mg,置100 mL量瓶中加稀释剂溶解并稀释至刻度,摇匀,作为固态下紫外光破坏溶液。
2.8.3 高温破坏取本品,置105 ℃烘箱中放置4 d。破坏结束,取高温破坏样品9.99 mg,置100 mL量瓶中,加稀释剂溶解并稀释至刻度,摇匀,作为高温破坏溶液。
2.8.4 氧化破坏取本品10.38 mg,精密称定,置100 mL量瓶中,加2 mL稀释剂,加10%过氧化氢2 mL,摇匀,90 ℃加热1 h,破坏结束后,加稀释剂溶解并稀释至刻度,摇匀,作为氧化破坏溶液。
2.8.5 酸性氧化破坏取本品10.33 mg,精密称定,置100 mL量瓶中,加2 mL稀释剂,加10%过氧化氢2 mL、6 mol·L-1盐酸溶液2 mL,摇匀,90 ℃加热3 h,破坏结束后,加6 mol·L-1氢氧化钠溶液2 mL中和,加稀释剂溶解并稀释至刻度,摇匀,作为酸性氧化破坏溶液。
2.8.6 酸破坏取本品10.08 mg,精密称定,置100 mL量瓶中,加稀释剂2 mL使溶解,加6 mol·L-1盐酸溶液2 mL,摇匀,置90 ℃水浴中放置12 h,破坏结束后,加6 mol·L-1氢氧化钠溶液2 mL中和,摇匀,加稀释剂溶解并稀释至刻度,摇匀,作为强酸破坏溶液。
2.8.7 碱破坏取本品10.32 mg,精密称定,置100 mL量瓶中,加稀释剂2 mL使溶解,加6 mol·L-1氢氧化钠溶液2 mL,摇匀,置90 ℃水浴中放置13 h,破坏结束后,加6 mol·L-1盐酸溶液2 mL,摇匀,加稀释剂溶解并稀释至刻度,摇匀,作为强碱破坏溶液。
取各破坏试验供试品溶液按“2.1”色谱条件进样测定,考察各破坏试验条件下降解杂质与已知杂质、主成分的分离情况,见图 2。咪喹莫特原料药经酸、碱、光照和高温破坏后,杂质含量无明显变化,表明本品在上述条件下比较稳定,咪喹莫特在氧化条件下特别在酸存在下不稳定,主成分含量从99.84%降至96.53%,DAD检测结果表明,各降解条件下主峰纯度因子均大于990,且各杂质均能与咪喹莫特主峰良好的分离,物料平衡均在98.31%~104.06%之间,符合专属性要求。

|
1.强白光破坏(light damage)2.紫外破坏(UV damage)3.高温破坏(heat damage)4.酸破坏(acid damage)5.碱破坏(base damage)6.氧化破坏(oxidative damage)7.酸性氧化破坏(acid oxidative damage) 图 2 咪喹莫特破坏试验高效液相色谱图 Figure 2 HPLC chromatograms of degradation tests of imiquimod |
按照“2.2.2”项下配制供试品溶液,分别于室温放置0 h、6 h、10 h、24 h后测定溶液稳定性,面积归一法计算主峰纯度分别为99.88%、99.89%、99.90%、99.90%,24 h内杂质没有增长,但供试品溶液放置6 h后杂质个数由6个减少至5个,为真实反映杂质的含量,故本品溶液应在6 h内检测。
2.10 重复性试验取同一批号供试品,按照“2.2.2”和“2.2.3”项下配制供试品溶液和对照溶液,并按照上述色谱条件进行测定,结果表明6份供试品中杂质A均为0.01%,RSD为0;杂质C均为0.03%,RSD为0;杂质D均为0.01%,RSD为0;杂质B和杂质E均未检出;其他单杂均为0.04%,RSD为0;杂质总量分别为0.12%、0.13%、0.13%、0.14%、0.13%、0.14%,RSD为5.7%,表明该方法重复性良好。
2.11 样品检测分别采用杂质对照品外标法与校正因子的主成分自身对照法,测定3批咪喹莫特原料药中各杂质的含量,结果见表 6。用加校正因子的主成分自身对照法和对照品外标法杂质的检测结果基本一致,无统计学差异,终点验证了校正因子的准确性。
|
|
表 5 重复性试验结果(%) Table 5 The results of repeatability test |
|
|
表 6 样品测定结果(%) Table 6 The results of sample analysis |
USP39收载的咪喹莫特标准和新药转正标准WS1-(X-125)-2005Z有关物质方法对已知杂质的分离均存在一定的缺陷,其中USP39收载的咪喹莫特标准有关物质方法杂质E与主成分相互干扰,不能实现完全分离,流动相含离子对试剂,对色谱柱伤害大,分析时间较长;新药转正标准WS1-(X-125)-2005Z有关物质方法咪喹莫特与相邻杂质的分离度达不到分离要求,其中咪喹莫特与杂质A的分离度只有0.89,咪喹莫特与杂质E与的分离度只有1.36。新药转正标准WS1-(X-125)-2005Z-2010中有关物质方法各已知杂质之间及与主成分之间均能完全分离,且流动相组成简单,分析时间较美国药典方法短,故咪喹莫特原料药有关物质选择WS1-(X-125)-2005Z-2010收载的方法。
3.2 稀释剂的选择由于咪喹莫特原料药在水和有机溶剂中溶解度均不佳[14-15],在酸性条件溶解性较好[16-17],稀释剂宜选择酸性溶液,考虑到杂质的溶解性,故选择0.02 mol·L-1磷酸溶液-乙腈(70:30)作为稀释剂。
3.3 流动相比例的选择减少乙腈比例有利于提高杂质B与未知相邻杂质的分离度,但却会降低杂质C与相邻杂质的分离度。综合考虑杂质B、杂质C与相邻杂质的分离度,通过多次调节流动相的组成和比例,最终确定以0.02 mol·L-1磷酸氢二钾缓冲液(用磷酸调节pH至8.0)-乙腈(70:30)为流动相,杂质B、杂质C与相邻杂质分离度达到最佳。
3.4 色谱柱的筛选本实验考察采用Agilent XDB-C18、Waters Sunfire C18、Waters XBridge C18等色谱柱对各杂质相对保留时间与分离度的影响,结果表明色谱柱品牌和型号对主峰出峰时间、已知杂质的相对保留时间、主峰与相邻杂质的分离度有显著影响,特别是杂质A的相对保留时间,甚至会引起杂质A与主成分出峰顺序的不同,Waters XBridge C18适合咪喹莫特有关物质的测定方法,且不同批次的色谱柱各已知杂质的相对保留时间与分离度一致,方法可重现。
3.5 相对保留时间考察取“2.4”项下分离度试验溶液在“2.1”色谱条件基础上微小变动,分别考察流速、柱温、流动相比例、流动相中磷酸氢二钾溶液的pH、色谱柱批次对杂质相对保留时间的影响,结果见表 7。改变流速、柱温、流动相比例、流动相pH、不同批次的色谱柱,各已知杂质的相对保留时间耐用性良好。
|
|
表 7 各种影响因素对相对保留时间的影响(n=10) Table 7 Effects of various factors on the relative retention time |
本文通过测定咪喹莫特杂质A、B、C、D和E的相对校正因子,建立加校正因子的主成分自身对照法测定咪喹莫特原料药的有关物质,解决杂质对照品不易获得的难题,同时避免不加校正因子的主成分自身对照法定量不准确的缺陷,本法测定结果与杂质对照品外标法测定的结果一致,终点验证校正因子的准确性。建立的方法进行了系统的方法学验证,结果表明该方法灵敏度、准确度、专属性、重复性、溶液稳定性均符合要求。采用加校正因子的主成分自身对照法测定咪喹莫特有关物质无需长期购买杂质对照品,大大降低了检验成本和检验人员的劳动程度,故本文建立的加校正因子的主成分自身对照法可用于咪喹莫特有关物质的测定,方法简单、专属性强、结果准确可靠。
| [1] |
王琨. 咪喹莫特的临床应用新进展[J]. 临床皮肤科杂志, 2016, 45(1): 72. WANG K. New advances in clinical application of imiquimod[J]. J Clin Dermatol, 2016, 45(1): 72. |
| [2] |
王强, 倪立燕, 姚学军. 外用咪喹莫特预防尖锐湿疣复发的疗效及其机制[J]. 中国临床医学, 2010, 17(6): 921 WANG Q, NI LY. Clinical observation on externally use dimiquimod cream 5% in preventing recurrence of condyloma acuminate and its pathogenesis, 2010, 17(6): 921 http://kns.cnki.net/KCMS/detail/detail.aspx?filename=lcyx201006061&dbname=CJFD&dbcode=CJFQ |
| [3] |
李剑峰, 章诗富. 咪喹莫特的免疫调节机制及临床应用进展[J]. 医学综述, 2009, 15(9): 1394. LI JF, ZHANG SF. Imunoregulatory in mechanism and the clinical application of imiquimod[J]. Med Recapit, 2009, 15(9): 1394. |
| [4] |
闫伦. 咪喹莫特对兔耳瘢痕模型增生抑制作用的实验研究[D]. 桂林: 桂林医学院, 2013 YAN L. Imiquimod Inhibit Scar Formation in Rabbit Ear Hypertrophic Scar Model[D]. Guilin: Guilin Medical University, 2013 http://cdmd.cnki.com.cn/Article/CDMD-10601-1013379491.htm |
| [5] |
朱小华, 徐金华. 5%咪喹莫特乳膏治疗基底细胞癌Meta分析[J]. 中国皮肤性病学杂志, 2006, 20(9): 568. ZHU XH, XU JH. Meta analysis of the effectiveness of 5% imiquimod in treatment of basal cell carcinoma[J]. Chin J Derm Venereol, 2006, 20(9): 568. |
| [6] |
新药转正标准: 第65册[S]. 2005: 170 New Positive Standard. Vol 65 [S]. 2005: 170 |
| [7] |
国家药品标准颁布件: 咪喹莫特[S]. 2010 National Drug Standard Issue Part: Imiquimod [S]. 2010 |
| [8] | |
| [9] |
美国药典论坛第一版: 咪喹莫特[S]. 2011 Authorized USP Pending Monograph Version 1: Imiquimod [S]. 2011 |
| [10] |
章雁, 程文香, 甘斌, 等. 咪喹莫特的合成[J]. 中国药师, 2010, 13(2): 209. ZHANG Y, CHENG WX, GAN B, et al. Synthesis of imiquimod[J]. China Pharm, 2010, 13(2): 209. |
| [11] |
张逸伟, 陈海权, 何伟彪, 等. 咪喹莫特合成工艺的改进[J]. 华南理工大学学报自然科学版, 2007, 35(7): 78. ZHANG YW, CHEN HQ, HE WB, et al. Improved synthesis of imiquimod[J]. J South China Univ Technol (Nat Sci Edit), 2007, 35(7): 78. |
| [12] |
沈敬山, 李剑峰, 李卉君, 等. 咪喹莫特的合成研究[J]. 化学研究与应用, 2001, 13(3): 249. SHEN JS, LI JF, LI HJ, et al. Synthesis research of imiquimod[J]. Chem Res Appl, 2001, 13(3): 249. |
| [13] |
夏广新, 张容霞, 索瑾, 等. 咪喹莫特的波谱学数据与结构表征[J]. 分析化学研究报告, 2003, 31(10): 1185. XIA GX, ZHANG RX, SUO J, et al. Spectroscopic data and structural characterization of imiquimod[J]. Chin J Anal Chem, 2003, 31(10): 1185. |
| [14] |
孙镜沂, 孙永超, 丁平田. 促渗剂对咪喹莫特体外经皮渗透的影响[J]. 沈阳药科大学学报, 2008, 25(2): 85. SUN JX, SUN YC, DING PT. Effects of penetration enhancers on the transdermal permeation of imiquimod[J]. J Shenyang Pharm Univ, 2008, 25(2): 85. |
| [15] |
许莉勇. 顶空气相色谱法测定咪喹莫特中有机溶剂残留量[J]. 药物分析杂志, 2010, 30(6): 1086. XU LY. Headspace GC determination of residual solvents in imiquimod[J]. Chin J Pharm Anal, 2010, 30(6): 1086. |
| [16] |
郑萍, 杜开蓉, 王舸. 反相高效液相色谱法测定咪喹莫特乳膏的有关物质及含量[J]. 中国抗生素杂志, 2004, 29(10): 600. ZHENG P, DU KR, WANG K. Determination of content and related substances in imiquimod cream by RP-HPLC[J]. Chin J Antibiot, 2004, 29(10): 600. |
| [17] |
曹琳, 周征, 毛亚珠. 高效液相色谱法测定咪喹莫特乳膏的含量[J]. 中国药品标准, 2008, 9(3): 227. CAO L, ZHOU Z, MAO YZ. Determination of imiquimod cream by HPLC[J]. Drug Stand China, 2008, 9(3): 227. |