 2017, Vol. 37
2017, Vol. 37 

注射用盐酸甲氯芬酯为苯氧乙酸酯类中枢兴奋药,用于治疗外伤性昏迷、酒精中毒、儿童遗尿症、意识障碍等[1-4]。工业化生产盐酸甲氯芬酯主要以对氯苯氧乙酸为起始原料,与N,N-二甲氨基乙醇反应生成甲氯芬酯,然后经过成盐、结晶制得[5-6]。根据企业调研结果,本次测定的66批注射用盐酸甲氯芬酯,其制剂工艺分为原料药直接分装和冷冻干燥2种,后者含有辅料甘露醇。盐酸甲氯芬酯原料药及注射用粉针质量标准收载于中国药典2015年版[7],有关物质检查采用高效液相色谱法对单个杂质和杂质总量进行控制;日本药局方收载其原料药标准[8],且无有关物质项检查。由于甲氯芬酯结构中含有酯键,极易水解,而现行质量标准并未对特定杂质进行控制,因此,为了更好地掌握该药物的杂质组成及特性,评价不同企业药品有关物质控制水平的高低,本文对其制剂杂质谱进行了相关研究,确定杂质结构及来源,特别是结合不同工艺考察其杂质谱的差异。
通常杂质谱研究包括杂质检测和杂质结构鉴定两方面内容[9-10],本文在对注射用盐酸甲氯芬酯进行杂质检测和杂质定向合成,确证其结构的基础上,进一步对其进行溯源分析,结合生产工艺推测论证其产生原因,并提出针对性防控措施,为提升注射用盐酸甲氯芬酯的药品质量,提供一种新的杂质谱研究思路。
1 仪器与试药 1.1 仪器Shimadzu 20A高效液相色谱仪(日本岛津公司),6510 Q-TOF四极杆-飞行时间质谱仪(美国Agilent公司),AVANCEIII 400兆核磁共振谱仪(美国Bruker公司),Nicolet 5700傅里叶红外光谱仪(美国Thermo Fisher公司),Thermo TSQ-Quantum气质联用仪(美国Thermo Fisher公司),Milli-Q纯水机(德国merck millipore公司),电子天平(德国赛多利斯天平公司)。
1.2 样品及试剂注射用盐酸甲氯芬酯样品涉及9家企业共66批(其中无菌分装样品44批,冷冻干燥样品22批),均为国家药品评价性抽验样品。盐酸甲氯芬酯对照品(批号100840-201402,含量99.7%)购自中国食品药品检定研究院;杂质对照品对氯苯氧乙酸(批号05621LT-1)购自北京百灵威科技有限公司,其余杂质对照品均由中国医学科学院药物研究所协助合成。辅料甘露醇(批号361504002)由青岛明月海藻集团有限公司提供。辛烷磺酸钠为分析纯,购自北京百灵威科技有限公司。乙腈为色谱纯,实验用水为超纯水,其他试剂均为分析纯。
2 方法与结果 2.1 杂质检测按照中国药典2015年版二部注射用盐酸甲氯芬酯有关物质的测定方法,采用资生堂MG-Ⅱ C18(4.6 mm×150 mm,5 μm;填料:十八烷基硅烷键合硅胶)色谱柱,流动相为0.05 mol·L-1辛烷磺酸钠溶液(磷酸调节pH至2.5)-乙腈(65:35),检测波长225 nm,根据样品中检出杂质的实际情况将记录时间延长至主成分保留时间的4倍。对66批样品进行检测,共发现3种杂质,典型色谱图见图 1,其中杂质A、C为高频出现的主要杂质,且所有样品均检测出杂质A,在14批样品中检测出杂质C,并在5批样品中检测出杂质B。经结构确证,杂质C与盐酸甲氯芬酯的合成工艺相关,且在国内外均未见报道。

|
1.盐酸甲氯芬酯(meclofenoxate hydrochloride)2.杂质A(impurity A)3.杂质B(impurity B)4.杂质C(impurity C) 图 1 注射用盐酸甲氯芬酯典型色谱图 Figure 1 HPLC chromatogram of meclofenoxate hydrochloride for injection |
对杂质A、B、C的含量进行分析汇总。杂质A均值为0.16%,最大值1.14%;杂质B均值为0.01,最大值0.02%;杂质C均值为0.11%,最大值0.01%。现行标准限度为单个杂质不得过0.5%,杂质总量不得过1.0%。有关物质不合格的3批样品均为杂质A含量超限,且均为冷冻干燥工艺生产的制剂。杂质C仅在1家企业中检出。
2.2 杂质结构确证现行中国药典标准中注射用盐酸甲氯芬酯有关物质采用主成分自身对照法,虽然在标准中提到了相对保留时间约为0.6的杂质峰,但并未明确杂质结构,亦未对其他主要杂质进行描述和定性分析。
采用液相色谱-飞行时间质谱(HPLC-TOF)联用技术对样品中可能含有的杂质进行了分析和结构确定。使用CAPCELL PAK CR 1:20色谱柱(2.0 mm×150 mm,5 μm),流动相为水(甲酸调节pH至2.5)-乙腈(65:35),流速为0.2 mL·min-1,柱温为30 ℃。电喷雾离子源(ESI),电压4.5 kV,毛细管温度350 ℃,碰撞电压120 V,鞘气流速40 arb,正离子模式,二级全扫描方式检测。应用质谱软件通过一级质谱数据获得分子式,并通过二级质谱数据进行结构确认,根据降解原理和工艺,推断出样品中3个杂质的结构(见表 1)。根据推断的杂质结构,由中国医学科学院药物研究所协助合成,获得了杂质对照品。通过红外光谱、核磁共振光谱、高分辨质谱对杂质进行鉴定,杂质A即为药典标准中提到的水解产物,杂质B和杂质C为醇解产物。对获得的杂质对照品采用国际通用的质量平衡法进行标化,以面积归一化法测得对照品的色谱纯度,并扣除对照品中含有的水分及杂质,色谱条件同“2.1杂质检测”方法,杂质对照品的纯度如表 1所示。
|
|
表 1 盐酸甲氯芬酯杂质结构 Table 1 Structure of impurities in meclofenoxate hydrochloride |
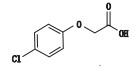
杂质A为对氯苯氧乙酸,既是盐酸甲氯芬酯的合成起始物料,又是其水解产物。经检测,冷冻干燥工艺样品中杂质A含量(均值0.34%)高于原料药,且约为无菌分装工艺样品中杂质A含量(均值0.05%)的7倍。说明样品中杂质A的主要来源为冻干工艺中水解产生。通过模拟生产工艺,考察了溶液pH值、配制时间和温度对水解反应的影响。
取1 mg·mL-1盐酸甲氯芬酯水溶液,临用新制,在室温(25 ℃)下分别用磷酸调pH值至3.0、3.5、4.0、4.5和5.0后立即进样检测,考察8 h内杂质A含量的变化。结果如图 2所示,杂质A的生成量随溶液pH值的增加而增加,当溶液pH大于4.0时,杂质A的生成量增长显著。

|
图 2 不同pH溶液中杂质A含量随时间变化曲线 Figure 2 Content curves of impurity A with time in different pH solutions |
按照含辅料最高比例的处方,取盐酸甲氯芬酯原料药和甘露醇按1:1.7的比例配制成含盐酸甲氯芬酯1 mg·mL-1的溶液(pH约为5),临用新制,分别溶解于5 ℃、10 ℃、15 ℃和20 ℃的注射用水中,立即进样检测,并保持样品温度分别为5 ℃、10 ℃、15 ℃和20 ℃,考察8 h内杂质A含量的变化,结果如图 3所示,杂质A的生成量随配制温度和时间的增加而增加。

|
图 3 不同温度溶液中杂质A含量随时间变化曲线 Figure 3 Content curves of impurity A with time in different temperature solutions |
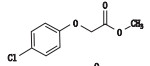
杂质B为对氯苯氧乙酸甲酯,是盐酸甲氯芬酯与甲醇发生酯交换反应的产物,仅在1家企业的样品中检出,含量为0.01%~0.03%。取1 mg·mL-1盐酸甲氯芬酯的甲醇溶液,立即检测,反应30 min后杂质B含量为1.0%,说明盐酸甲氯芬酯在甲醇溶液中会迅速降解,产生大量的杂质B。根据企业提供的生产工艺分析,检出杂质B的企业所用原料药在合成中使用了大量的异丙醇,推测可能是所用的异丙醇纯度较低,其中所含的少量甲醇与盐酸甲氯芬酯发生了醇解反应,产生杂质B。实验证实,将盐酸甲氯芬酯置于纯度为99.9%的异丙醇溶液中,反应24 h,产生杂质B的含量为0.03%。
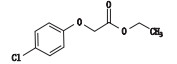
2.3.3 杂质C的来源分析及与工艺相关性杂质C为对氯苯氧乙酸乙酯,是盐酸甲氯芬酯与乙醇发生酯交换反应的产物,仅在1家企业的样品中检出,含量为0.1%~0.2%。经调研发现,各企业虽然在原料药的精制过程中均用到了无水乙醇,但只有该企业的原料药在使用无水乙醇的同时加入了冰醋酸。
采用气质联用法测定所有企业样品中的残留溶剂[11],仅在检出杂质C的企业样品中,检测到了乙醇(含量0.1%~0.2%)和乙酸乙酯(含量0.1%~0.2%),其中乙酸乙酯为乙醇和冰醋酸反应的产物。由此推断杂质C在原料药合成工艺中引入,为酸性条件下甲氯芬酯与无水乙醇的酯交换产物,冰醋酸作为酯交换反应的催化剂,加速了杂质C的生成。
3 讨论 3.1 色谱条件的优化在现行中国药典标准的有关物质色谱条件下,盐酸甲氯芬酯与各杂质能有效分离,但在低有机相比例等度洗脱条件下分析时间较短(标准规定为主成分保留时间的2倍),可能有强保留杂质未能检出。适当延长洗脱时间后,发现个别企业样品在相对保留时间约为3.0时有一较大色谱峰被洗脱(杂质C),如图 1所示。进一步实验证实,提高有机相比例及采用梯度洗脱的方法,均未有除杂质C外的其他新杂质检出。虽然梯度洗脱可以获得较高的理论板数,但流动相中使用了离子对试剂,需要较长的平衡时间。在等度洗脱中通过提高有机相比例可使杂质C在规定的分析时间内检出,但在专属性研究中氧化破坏的样品主峰与相邻杂质峰难以分离。因此,兼顾分离度要求,最终确定在中国药典有关物质色谱系统的基础上(有机相比例35%等度洗脱),通过延长分析时间至主成分保留时间的4倍来解决杂质C未能洗脱的问题。
3.2 杂质的防控措施盐酸甲氯芬酯的杂质谱研究表明,其杂质为水解杂质(杂质A)和醇解杂质(杂质B、杂质C)两种。由研究结果可知,水解杂质主要为制剂冷冻干燥生产工艺中引入,与配制过程中的溶液pH,配制温度和时间具有正相关。醇解杂质主要为原料药合成工艺中引入,与工艺中使用的低级醇类有关。因此,为了有效控制样品中杂质含量,建议在冻干制剂的配制过程中溶液pH不超过4.0,配制温度在10 ℃以下,并严格控制配制时间;在原料药精制纯化过程中应避免使用低级醇类试剂,且使用无水乙醇做溶剂时,应合理控制生产工艺以减少杂质C的生成。尤其是在有冰醋酸的酸性条件下,会加速甲氯芬酯与无水乙醇的酯交换反应[12],推断其反应机理如图 4所示。

|
图 4 酸性条件加速甲氯芬酯与乙醇的反应机理 Figure 4 Reaction mechanism of meclofenoxate and alcohol accelerated in acidity condition |
本文研究了盐酸甲氯芬酯杂质谱的检测和构成,并通过定向合成验证杂质结构,其中杂质A是主要降解产物,需重点控制。本实验的研究方法为完善中国药典2015年版二部中盐酸甲氯芬酯原料及制剂的质量标准提供了参考,研究结果合理解释了有关物质不合格样品的杂质成因。通过分析杂质来源,为改进工艺提供依据,对提高产品质量具有重要意义。
| [1] |
吕卫红. 盐酸甲氯芬酯的药理作用和临床应用[J]. 西北药学杂志, 2004, 19(1): 47. LU WH. The pharmacologic action and clinical application of meclofenoxate hydrochloride[J]. Northwest Pharm J, 2004, 19(1): 47. |
| [2] |
周桂花, 汪萌芽. 药物治疗酒精中毒的研究进展[J]. 中国当代医药, 2013, 20(16): 22. ZHOU GH, WANG MY. Research advances in drug therapy of alcohol poisoning[J]. China Mod Med, 2013, 20(16): 22. DOI:10.3969/j.issn.1674-4721.2013.16.009 |
| [3] |
蒲友华, 刘定远. 注射用盐酸甲氯芬酯治疗新生儿缺氧缺血性脑病临床研究[J]. 儿科药学杂志, 2008, 14(4): 47. PU YH, LIU DY. The clinical research of meclofenoxate in the treatment of neonatal hypoxic-ischemic encephalopathy[J]. J Pediat Pharm, 2008, 14(4): 47. |
| [4] |
张天锡. 注射用盐酸甲氯芬酯的临床应用[J]. 药学服务与研究, 2005, 5(4): 388. ZHANG TX. Clinical application of meclofenoxate hydrochloride injection[J]. Pharm Care Res, 2005, 5(4): 388. |
| [5] |
赵普. 盐酸甲氯芬酯的合成[J]. 齐鲁药事, 2012, 31(12): 690. ZHAO P. Synthesis of meclofenoxate hydrochloride[J]. Qilu Pharm Aff, 2012, 31(12): 690. |
| [6] |
张忠敏, 张利敏, 修荣, 等. 氯醒酯的合成工艺改进[J]. 中国药物化学杂志, 2004, 2(14): 112. ZHANG ZM, ZHANG LM, XIU R, et al. Improved synthesis of meclofenoxatum[J]. Chin J Med Chem, 2004, 2(14): 112. |
| [7] |
中国药典2015年版. 二部[S]. 2015: 904 ChP2015. Vol Ⅱ[S]. 2015: 904 |
| [8] |
JP ⅩⅥ, 2011: 1065
|
| [9] |
胡昌勤. 化学药品杂质谱控制的现状与展望[J]. 中国新药杂志, 2015, 24(15): 1727. HU CQ. Current situation and the trend in impurity profiling of chemical drugs[J]. Chin J New Drugs, 2015, 24(15): 1727. |
| [10] |
郝杰, 张哲峰, 毕小平. 原料药杂质研究与控制浅析[J]. 中国现代应用药学, 2015, 32(6): 757. HAO J, ZHANG ZF, BI XP. Analysis of method for testing and controlling the impurity of crude drug[J]. Chin J Mod Appl Pharm, 2015, 32(6): 757. |
| [11] |
吴兆伟, 陈安东, 王铁松, 等. 气相色谱-质谱法同时分析固体药物制剂中50种残留溶剂[J]. 中国药学杂志, 2014, 49(9): 764. WU ZW, CHEN AD, WANG TS, et al. Simultaneous determination of 50 residual solvents in solid drug preparations by GC-MS[J]. Chin Pharm J, 2014, 49(9): 764. |
| [12] |
杨振强, 谢文磊, 李海涛. 酸催化油脂酯交换反应研究进展[J]. 粮食与油脂, 2006(3): 13. YANG ZQ, XIE WL, LI HT. Research progress on transesterification of oils and fats via acid catalysts[J]. Cereals Oils, 2006(3): 13. |