 2017, Vol. 37
2017, Vol. 37 
酒石酸唑吡坦(zolpidem tartrate)是第1个新型非苯类镇静安眠药。酒石酸唑吡坦由两分子的唑吡坦和一分子的酒石酸成盐而成,相对分子质量为764.88,中文化学名为N,N,6-三甲基-2-(4-甲基苯基)-咪唑并[1,2-a]吡啶-3-乙酰胺-L-(+)-酒石酸盐,于1988年上市,1998年SFDA批准其在中国上市,商品名为思诺思[1]。通过选择性地与中枢神经系统的ω1-受体结合,产生药理作用[2],主要用于失眠症的短期治疗。文献报道,不良反应有遗忘、幻觉和精神障碍等[3]。
酒石酸唑吡坦属于第2类精神药品,随着国产酒石酸唑吡坦片的广泛使用,其质量问题也应引起关注。
2014年湖南省药品检验研究院承担了酒石酸唑吡坦片的国家计划抽验任务,抽验样品涉及4家生产企业23个批次,占生产企业数的50.0%。按执行标准测定有关物质结果显示,4个生产企业23批次样品最大单个杂质在0.01%~0.18%范围内,国产酒石酸唑吡坦片与参比产品(思诺思)具有不同的杂质谱,而且国产样品中最大单个杂质有随贮存时间延长而增加的趋势。国外药典对酒石酸唑吡坦片均有控制特殊杂质和其他单个杂质,USP 39规定特殊杂质和其他单个杂质均不得过0.3%,进口药品注册标准JX20100222规定特殊杂质和其他单个杂质均不得过0.2%,中国药典2015年版二部仅规定了杂质总量不得过0.5%。由于国产药品的执行标准均未控制单个杂质的量,而测定结果表明个别杂质量大于0.1%,且随贮存时间延长而增加,这可能与国产药品的处方工艺有关。
查阅国内外文献,涉及此类药品的文献多为不良反应[4]、含量测定[5-7]、药代动力学研究[8-9]以及合成工艺[10],杂质研究仅有关于思诺思的5个杂质(m/z 253、457、354、294和295)结构的报道[11],然而国产酒石酸唑吡坦片的杂质研究尚未见报道。目前国内的生产企业获得批文的时间较早,各生产企业均使用本企业生产的原料制成制剂,由于工艺路线不同,杂质控制是保证酒石酸唑吡坦质量的关键要素。按照中国药典2015年版四部药品杂质分析指导原则[12],本文采用Bruker impact Ⅱ超高分辨飞行时间质谱结合SmartFormula软件对酒石酸唑吡坦的杂质进行分析鉴定。本文的研究有助于对酒石酸唑吡坦处方工艺进行有效监控,保证药品生产流程的质量稳定性,可为酒石酸唑吡坦原料药及其制剂的质量控制提供参考。
1 仪器与试药Agilent1260型高效液相色谱仪,Bruker impact Ⅱ超高分辨飞行时间质谱,SmartFormula软件(由布鲁克公司开发,通过测定化合物的真实同位素峰形预测准确分子式)。
酒石酸唑吡坦对照品(中国食品药品检定研究院,批号171258-200601,105 ℃干燥2 h后使用),酒石酸唑吡坦片(A企业,批号111201;B企业,批号140201),甲醇、乙腈为HPLC级,乙酸铵为AR级,水为Milli-Q水,其余试剂均为分析纯。
2 方法与结果 2.1 色谱-质谱条件 2.1.1 色谱条件色谱柱:Thermo Hypersil GOLD C18(2.1 mm×100 mm,1.9 μm);流动相:10 mmol·L-1乙酸铵溶液-甲醇-乙腈(53:23:18);流速:0.25 mL·min-1;柱温:30 ℃;进样量:5 μL。
2.1.2 质谱条件ESI离子源;离子化方式:正离子化;毛细管电压:4.5 kV;喷雾压力:200 kPa;干燥气流速:8.0 L·min-1;碰撞电压:7.0 eV;扫描方式:多级全扫描。
2.2 溶液配制 2.2.1 对照品溶液精密称取酒石酸唑吡坦对照品约5 mg,置10 mL量瓶中,加流动相超声溶解并定容,摇匀,配成质量浓度为0.5 mg·mL-1的溶液,即得。
2.2.2 供试品溶液精密称取样品细粉约60 mg,置10 mL量瓶中,加流动相超声(1 600 W,40 kHz)溶解并定容,摇匀,滤过,取续滤液即得。
2.2.3 氧化破坏溶液精密称取样品细粉约60 mg,置10 mL量瓶中,加3%过氧化氢溶液5 mL,室温放置1 h,用流动相稀释至刻度,摇匀,滤过,取续滤液即得。
2.2.4 光照破坏溶液精密称取强光(5 000 lx)照射5 d的样品细粉约60 mg,置10 mL量瓶中,加流动相超声溶解并定容,摇匀,滤过,取续滤液即得。
2.2.5 碱破坏溶液精密称取样品细粉约60 mg,置10 mL量瓶中,加1 mol·L-1氢氧化钠溶液2 mL,室温放置1 h,再加1 mol·L-1盐酸溶液调节pH至中性后,用流动相稀释至刻度,摇匀,滤过,取续滤液即得。
2.2.6 酸破坏溶液精密称取样品细粉约60 mg,置10 mL量瓶中,加1 mol·L-1盐酸溶液2 mL,室温放置1 h,再加1 mol·L-1氢氧化钠溶液调节pH至中性后,用流动相稀释至刻度,摇匀,滤过,取续滤液即得。
2.2.7 高温破坏溶液取供试品溶液置90 ℃水浴加热1 h,放冷,滤过,取续滤液即得。
2.3 唑吡坦的质谱解析取“2.2.1”项对照品溶液在“2.1”项条件下进样分析,鉴别唑吡坦的结构,得到唑吡坦的二级质谱图和相关裂解途径见图 1。可见m/z 308.176 8,为唑吡坦的[M+H]+离子,主要碎片离子为m/z 263、m/z 235和m/z 221。查阅文献,推测唑吡坦可能的裂解规律主要为侧链酰胺C-N键的断裂得到碎片m/z 263;随后脱掉一分子一氧化碳得到碎片m/z 235;进一步脱掉1个甲基,得到碎片m/z 221。

|
图 1 唑吡坦质谱图及裂解途径 Figure 1 Mass spectrogram and fragmentation pathway of zolpidem |
取“2.2.2”项供试品溶液,在“2.1”项条件下进样分析,得到酒石酸唑吡坦片杂质TIC图(见图 2)。

|
图 2 酒石酸唑吡坦对照品与酒石酸唑吡坦片杂质TIC图 Figure 2 TIC of zolpidem and impurities in zolpidem tartrate tablets |
取“2.2.3”项至“2.2.7”项酒石酸唑吡坦片的氧化、光照、碱、酸和高温破坏溶液,在“2.1”项条件下进样分析,各降解试验TIC图见图 3。

|
图 3 氧化、酸、高温、碱、光照降解试验TIC图 Figure 3 TIC of zolpidem under stress condition including oxidation, acid, heat, alakli and highlight |
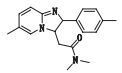
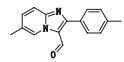
本文采用UHR-TOF/MS得到酒石酸唑吡坦片杂质的多级质谱图,除掉辅料峰共检测到杂质峰8个,使用SmartFormula软件预测准确分子式,结合酒石酸唑吡坦片的处方工艺和强降解试验条件,根据唑吡坦对照品的裂解规律,对杂质的裂解途径进行推测,初步鉴定了6个杂质的结构。峰6和峰8分别为USP 39收载的酒石酸唑吡坦片质量标准中有关物质项下杂质b(N,N-二甲基-2-(6-甲基-2-对甲苯基-咪唑并[1,2-a]吡啶-3-基)-2-氧代-乙酰胺)和杂质d(6-甲基-2-对甲苯基-咪唑并[1,2-a]吡啶-3-甲醛),其余均为未知杂质。峰2和峰3对应的杂质结构有待借助其他手段进一步分析鉴定。
杂质1(峰1)的准分子离子峰为366.183 4,为其[M+H]+峰,拟合元素组成,其离子式为C21H24N3O3,因而可知其分子式为C21H23N3O3,相比唑吡坦结构(C19H21N3O),多了片段C2H2O2。从唑吡坦的裂解规律中,得知了m/z 235的结构(见图 1),杂质1中因有m/z 235碎片,可知其与唑吡坦拥有相同的芳香区结构;而m/z 249碎片相比m/z 235碎片,m/z相差14,应为1个甲基,从而可推断出m/z 249碎片的结构;m/z 293碎片相比m/z 249碎片,m/z多了45,应为1个羧基,从而亦可得到m/z 293碎片的结构,结合唑吡坦的裂解规律,进而最终推断出杂质1的结构。对于该杂质的产生原因,推断是源于唑吡坦侧链的α氢有酸性,α氢被拔掉后生成的碳负离子具有亲核性,可与处方中羧甲淀粉钠发生取代反应。其结构及质谱裂解途径见图 4。

|
图 4 杂质1的质谱图及质谱裂解途径 Figure 4 Mass spectrogram and fragmentation pathway of impurity 1 |
杂质4(峰4)的准分子离子峰为294.160 4,为其[M+H]+峰拟合元素组成,其离子式为C18H20N3O,因而可知其分子式为C18H19N3O,相比唑吡坦结构(C19H21N3O),少了片段CH2。从二级质谱图中,发现该杂质具有与唑吡坦一致的裂解碎片。m/z 235以及m/z 221碎片峰的存在,可知其具有与唑吡坦相同的芳香区结构,从而可以推断出丢失的这个甲基应为酰胺氮的1个甲基。其结构及质谱裂解途径见图 5。

|
图 5 杂质4的质谱图及质谱裂解途径 Figure 5 Mass spectrogram and fragmentation pathway of impurity 4 |
杂质5(峰5)的准分子离子峰为324.170 2,为其[M+H]+峰拟合元素组成,其离子式为C19H22N3O2,从而得出其分子式为C19H21N3O2,与唑吡坦相比多了1个羟基(-OH)。因其碎片峰m/z 237与唑吡坦碎片峰m/z 235接近,因此两者应具有相同的芳香区,羟基应该连在唑吡坦侧链的α碳上。该杂质产生于唑吡坦的氧化。其结构及质谱裂解途径见图 6。

|
图 6 杂质5的质谱图及质谱裂解途径 Figure 6 Mass spectrogram and fragmentation pathway of impurity 5 |
杂质6(峰6)的准分子离子峰为322.155 0,为其[M+H]+峰拟合元素组成,其离子式为C19H19N3O2,可得其分子式为C19H18N3O2,与USP中记载的杂质b一致,来源于杂质3的进一步氧化。其结构及质谱裂解途径见图 7。

|
图 7 杂质6的质谱图及质谱裂解途径 Figure 7 Mass spectrogram and fragmentation pathway of impurity 6 |
杂质7(峰7)的准分子离子峰为310.191 3,为其[M+H]+峰,拟合元素组成,其离子式为C19H23N3O,可得其分子式为C19H22N3O,与唑吡坦分子式(C19H21N3O)比较,多了2个H;且其产生的碎片m/z 237、223同样比唑吡坦的碎片m/z 235、221多2个H,据此可知多出的2个H在芳香区。其结构及质谱裂解途径见图 8。

|
图 8 杂质7的质谱图及质谱裂解途径 Figure 8 Mass-spectrogram and fragmentation pathway of impurity 7 |
杂质8(峰8)的准分子离子峰为251.117 5,为其[M+H]+峰拟合元素组成,其离子式为C16H15N2O,可得其分子式为C16H14N2O。除去唑吡坦母核6-甲基-2-(4-甲基苯基)咪唑吡啶C15H14N2,剩下的CHO应为一醛基,结合USP中记载的杂质d结构,推测该杂质为唑吡坦甲醛。其结构及质谱裂解途径见图 9。

|
图 9 杂质8的质谱图及质谱裂解途径 Figure 9 Mass-spectrogram and fragmentation pathway of impurity 8 |
酒石酸唑吡坦片的杂质含量很低,主成分唑吡坦峰对称性对杂质的检出影响较大,色谱柱的选择非常关键[13]。各国药典和进口药品注册标准JX20100222收载的酒石酸唑吡坦有关物质方法均采用C18柱,美国药典的L1柱实际上也是C18柱,采用“2.1.1”项色谱条件对不同品牌不同粒径的色谱柱进行筛选,结果表明,采用Thermo-C18(2.1 mm×100 mm,1.9 μm),主峰对称性好,杂质峰分离良好。
3.1.2 流动相的选择各国药典规定的酒石酸唑吡坦片有关物质检查方法项下的流动相均含有大量的盐,不适用于本文的质谱检测,因此考察不同浓度不同pH的乙酸铵溶液,调节各种比例的甲醇和乙腈,结果表明流动相中加入适量的乙酸铵,唑吡坦峰的对称性好,理论板数高,与相邻杂质峰分离度良好[14]。
3.2 强降解试验条件的优化酒石酸唑吡坦的化学性质比较稳定[15],较温和的破坏试验条件对唑吡坦杂质的影响不明显,因此酸碱破坏均采用1 mol·L-1盐酸溶液和氢氧化钠溶液,破坏时间均为1 h,杂质1在酸性、碱性、光照和氧化条件下降解较大,在高温条件下略有增加,光照条件下产生杂质8。根据杂质在不同破坏条件下峰面积的变化情况对杂质可能产生的来源进行了分析,结果见表 1。
|
|
表 1 酒石酸唑吡坦片的杂质谱 Table 1 Impurity profile of zolpidem tartrate tablets |
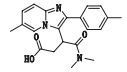
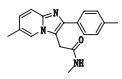
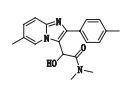
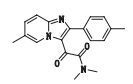
与传统的液质联用系统相比,UHR-TOF/MS不仅可以获得精确的质核比,而且可以确保测定得到化合物的真实同位素分布模式(TIP),TIP可以作为计算分子式的第2个参数即mSigma值。该值越小,说明实际测定的同位素分布模式与理论计算的同位素分布模式愈接近。SmartFormula软件在确定化合物的分子式时,不仅仅考虑精确质量的测定结果,更重要的是同时考虑同位素峰型的误差,对得到的多个可能的分子式,使用mSigma值确定准确分子式。
UHR-TOF/MS技术结合SmartFormula软件的应用缩小了分子式选择的范围,大大减少了后续结构解析的工作量,为快速准确的杂质鉴定提供了技术支持。
| [1] |
金永寿. 镇静催眠药思诺思[J]. 国外医药, 2000, 21(2): 128. JIN YS. Hypnotic drugs of stilnox[J]. World Pharm, 2000, 21(2): 128. |
| [2] |
刘曙晨. 镇静催眠药的研究进展[J]. 国外医学药学分册, 2000, 27(4): 227. LIU SC. Recent progress in development of hypnotic drugs[J]. Foreign Med Sci Sect Pharm, 2000, 27(4): 227. |
| [3] |
刘阳. 唑吡坦的不良反应[J]. 临床荟萃, 2008, 23(22): 1670. LIU Y. Side Effects of zolpidem[J]. Clin Focus, 2008, 23(22): 1670. |
| [4] |
UEMURA SI, KANBAYASHI T, WAKASA M, et al. Residual effects of zolpidem, triazolam, rilmazafone and placebo in healthy elderly subjects:a randomized double-blind study[J]. Sleep Med, 2015, 16(11): 1395. DOI:10.1016/j.sleep.2015.05.021 |
| [5] |
REZAEE ZAVAREH E, KIANI A, SHEIKHOLESLAM Z, et al. A Rapid HPLC method for determination of zolpidem and its degradation product in tablets using a monolithic column[J]. J Chromatogr Sci, 2015, 53(5): 807. DOI:10.1093/chromsci/bmu129 |
| [6] |
KHALDUN M, AL AZZAM, LEE KAM YIT, et al. Development and validation of a stability-indicating capillary electrophoresis method for the determination of zolpidem tartrate in tablet dosage form with positive confirmation using 2D-and 3D-DAD fingerprints[J]. Sci Pharm, 2014, 82(2): 341. DOI:10.3797/(ISSN)0036-8709 |
| [7] |
NEZAMI RASHID SAEED, YARIPOUR SAEID, LIMOHAMmadi A. A validated stability-indicating HPLC method for the quality control of zolpidem in pharmaceutical dosage forms[J]. Pharm Sci, 2014, 20(Supp 1): 53. |
| [8] |
朱定姬, 黄克建, 林翠梧, 等. 固相萃取-高效液相色谱法对血液中唑吡坦及其两种羧酸代谢物的同时测定[J]. 分析测试学报, 2010, 29(1): 22. ZHU DJ, HUANG KJ, LIN CW, et al. Simultaneous determination of zolpidem and its two carboxyl metabolites in blood by high performance liquid chromatography with solid phase extraction[J]. J Instrum Anal, 2010, 29(1): 22. |
| [9] |
SHI Y, XIANG P, SHEN BH, et al. A rapid and accurate UPLC/MS/MS method for the simultaneous determination of zolpidem and its main metabolites in biological fluids and its application in a forensic context[J]. J Chromatogr B Anal Technol Biomed Life Sci, 2012, 911: 140. DOI:10.1016/j.jchromb.2012.10.018 |
| [10] |
SUMALATHA Y, REDDY TR, PADI PR, et al. A simple, efficient and scalable synthesis of hypnotic agent, zolpidem[J]. ARKIVOC, 2009(ii): 315. |
| [11] |
SUMALATHA Y, PADI PR, REDDY R, et al. Synthesis and spectral characterization of process-related substances to the hypnotic agent zolpidem[J]. ARKIVOC, 2009(vii): 143. |
| [12] |
中国药典2015年版. 四部[S]. 2015: 377 ChP 2015. Vol Ⅳ[S]. 2015:377 |
| [13] |
YARIPOUR S, RASHID SN, ALIBAKHSHI H, et al. Development and validation of a stability-indicating reversed phase HPLC method for the quality control of zolpidem in bulk and tablet dosage forms[J]. J Anal Chem, 2015, 70(6): 738. DOI:10.1134/S1061934815060143 |
| [14] |
ELIASSEN E, KRISTOFFERSEN L. Quantitative determination of zopiclone and zolpidem in whole blood by liquid-liquid extraction and UHPLC-MS/MS[J]. J Chromatogr B, 2014, 971: 72. DOI:10.1016/j.jchromb.2014.09.018 |
| [15] |
KIM J, LEE S, IN S, et al. Validation of a simultaneous analytical method for the detection of 27 enzodiazepines and metabolites and zolpidem in hair using LC-MS/MS and its application to human and rat hair[J]. J Chromatogr B, 2011, 879(13): 878. |