 2017, Vol. 63
2017, Vol. 63
文章信息
- 朱华, 郑凡, 祝佳丽, 汪的华
- ZHU Hua, ZHENG Fan, ZHU Jiali, WANG Dihua
- 几种添加剂对锰沉积过程的作用机理
- Roles of Several Additives in the Manganese Electrodeposition
- 武汉大学学报(理学版), 2017, 63(1): 75-80
- Journal of Wuhan University(Natural Science Edition), 2017, 63(1): 75-80
- http://dx.doi.org/10.14188/j.1671-8836.2017.01.010
-
文章历史
- 收稿日期:2016-06-22



引用本文

|


1920年,英国的Allmand和Campbell[1]使用陶瓷隔膜制备得到了高纯度的电解锰,随后,美国矿山局通过硫酸浸取碳酸锰矿石得到硫酸锰电解质,并以SO2作为电解添加剂,采用纤维布袋替代陶瓷隔膜,以低的电流密度,恒电流电解48 h得到电解锰,从此,该工艺被广泛采用[2~4].
在电解锰生产过程中,存在着两个竞争的阴极反应:
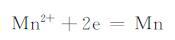
|
(1) |
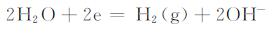
|
(2) |
由于氢的析出反应电位明显正于锰的沉积电位,意味着氢的析出反应难以避免,这也正是导致电解锰行业能耗高的根本原因.因此,为了提高锰的生产效率、降低能耗,必须尽可能抑制析氢反应.
Tilak等的研究发现[5],MnSO4-(NH4)2SO4-NH3\5H2O组成的电解质体系对Mn2+有较好的缓冲能力和络合能力,采用该体系可将Mn2+稳定存在的pH值提高至8左右.为了提高体系电解质的电导率,多选择高浓度的(NH4)2SO4作为支持电解质[6].有研究认为[7],添加SeO2和(NH4)2SO3能促进沉积的γ-Mn向导电性更高、更稳定的α-Mn转变,从而提高沉积效率.也有研究认为[8],SeO2具有明显的抑制析氢的作用,从而也可以提高锰沉积的效率.因此,深入研究现有添加剂在锰沉积过程的作用机理和电化学反应动力学特征,不仅有利于现有工艺的节能降耗,还可为新型添加剂的筛选和探索研究提供科学指导.
为此,本文首先采用电化学石英晶体微天平(electrochemical quartz crystal microbalance,EQCM)技术[9],精准地确认电解锰还原过程中锰沉积和析氢的电位范围,并在此基础上,采用三电极体系和单组分测试及组分分步添加的方式,通过循环伏安法(cyclic voltammetry,CV)比较电解质及添加剂对析氢和锰沉积的影响及其作用机制.所选添加剂浓度参考工业用量
1 实验部分 1.1 实验装置采用自制的有机玻璃电解槽,用阴离子交换膜将阴极室和阳极室分开,可以避免阳极室中氧化反应对阴极反应的干扰.单槽的规格为:8.5 cm×10 cm×5 cm,该电解槽可同时进行电沉积实验和三电极电化学测试.进行电化学测试时,将参比电极和工作电极布置在同一极室,构成三电极体系,进行测试即可.所有测试温度均保持在40±2 ℃.
1.2 电极制备1) 不锈钢盘电极:将市售304不锈钢棒(∅=0.8 cm,S=0.502 4 cm2)切割成5 mm长圆柱,一端用锡焊引出铜线做导线,整体以环氧树脂密封,经过打磨,露出不锈钢圆柱另一盘面作为研究电极.
2) 钛基涂层电极:Ru-Ir-Ta-Ti (3 cm×10 cm)涂层板封装后,背面绝缘,正面露出3 cm×3 cm的工作面.
3) 镀金石英晶振电极:基频9 MHz,∅=0.5 cm,石英晶体的密度为2.684 g/cm3 (25 ℃),石英晶体的剪切模数为2 948×1011 g/(m·s2).
1.3 电化学测试 1.3.1 EQCM测试采用Ametek公司的QCM922型石英晶体微天平与2273型电化学工作站联用,选用三电极体系,工作电极为镀金石英晶振电极,参比电极为饱和甘汞电极,对电极为钛基涂层阳极.采用循环伏安法,测试电势范围-0.8~-1.8 V,扫描速度5 mV\5s-1,阴极电解液(参考工业生产电解液配方):15 g\5L-1 Mn2+,150 g\5L-1 (NH4)2SO4,0.1 mol\5L-1 NH3\5H2O,0.2 g\5L-1 (NH4)2SO3(以SO2计),阳极电解液150 g\5L-1 (NH4)2SO4,调节pH值为7.0左右.
1.3.2 其他电化学测试同样采用三电极体系,除了工作电极选用304镜面不锈钢盘电极为工作电极外,其他同1.2节1),所用电解液见文中所述.
1.4 锰沉积实验及材料表征为了比较(NH4)2SO3和SeO2对锰沉积的影响和作用机理,采用上述电解槽,阴极为304镜面不锈钢板(4 cm×2 cm),阳极同1.2节2),工作电流280 mA,沉积2.5 h.电解液同1.3.1 节,添加剂及用量:0.2 g\5L-1 (NH4)2SO3(以SO2计)或0.03 g\5L-1 SeO2.
沉积锰的形貌表征采用Quanta200型扫描电子显微镜(SEM,FEI公司)进行;结构采用XRD-6000型X射线衍射仪(XRD,岛津公司)测试,测试条件:CuKα(λ=0.145 6 nm)为测试激发源,以连续扫描方式进行采样,扫描速度4(°)/min-1,扫描区间10°~80°,阶宽0.02°(2θ).
2 结果与讨论 2.1 EQCM分析EQCM能够在进行电化学极化的同时监测电极表面ng级的物质变化[10].图 1为模拟工业锰电解液的CV曲线及同步的质量变化(Δm)曲线.其中,阴极电流归属于Mn2+和H+在电极表面得到电子发生还原反应所产生的电流,阳极电流归属于沉积锰溶出所产生的氧化电流;Δm为Mn2+还原和沉积锰再溶出所产生的质量变化.从图 1中可以看出,负向扫描时,从-1.20 V至-1.60 V,电流缓慢增大,而Δm无显著变化,可见此电位区间主要的还原电流来自析氢反应;从-1.60 V开始,还原电流迅速增大,Δm也相应迅速增大,表明锰的起始沉积电位为-1.60 V;继续负向扫描,伴随着还原电流的增大,Δm持续增大,直至正向扫描至-1.60 V;然后,在-1.60 V~-1.48 V间出现了一质量变化的平台,说明该电势范围区间内,锰沉积和溶解的速度均较低,电极表面的表观质量维持不变;当正向扫描至大于-1.48 V后,伴随着氧化电流的快速增大,Δm快速降低,意味着电极表面沉积的锰开始发生溶解,质量变少.EQCM结果表明,析氢反应和锰沉积的起始电位分别为-1.20 V和-1.60 V.

|
| 图 1 模拟工业电解液的CV曲线及同步测试的Δm-E曲线 Figure 1 CV curve and the corresponding quality-potentials curve in the industrial electrolyte |
首先研究(NH4)2SO4电解体系中各种添加剂的作用.工业电解锰采用NH3\5H2O作为添加剂是基于氨水与Mn2+的络合作用.25 ℃时,当pH≥7.0时,Mn2+易水解沉淀,而添加的氨水因与Mn2+形成络合离子而使溶液中Mn2+能稳定存在的溶液pH值提高至8.0,但过量的氨水则会导致氨气溢出造成浪费和污染.为此,本研究选择测试电解液为150 g\5L-1 (NH4)2SO4溶液,分别添加0.1 mol\5L-1 NH3\5H2O(pH 7.6)和0.2 mol\5L-1 NH3\5H2O(pH 8.0)以及未添加NH3\5H2O(采用NaOH溶液调节pH值为7.6)3种体系,CV测试结果见图 2.同时,在上述实验基础上,考察添加剂(NH4)2SO3(添加量为0.2 g\5L-1,以SO2计)及SeO2(添加量为0.03 g\5L-1)对析氢反应的影响,结果见图 3.

|
| 图 2 不同浓度氨水-(NH4)2SO4体系的CV曲线 Figure 2 CV curves of (NH4)2SO4 system with different concentration of NH3\5H2O |

|
| 图 3 不同添加剂150 g\5L-1 (NH4)2SO4+0.1mol \5L-1NH3\5H2O体系的CV曲线 Figure 3 CV curves of 150 g\5L-1 (NH4)2SO4 +0.1 mol\5 L-1 NH3\5H2Osystem with different additives |
从图 2中可见,随着扫描电位负移,3种条件下的阴极电流均快速增大,对应H+在阴极得到电子生成氢气的还原反应.由于氢气生成后即从阴极表面脱离进入到电解质溶液中,故未见明显的氧化电流.当加入不同浓度的NH3\5H2O时,电极表面的还原电流均变小,说明NH3\5H2O的加入抑制了析氢反应.由于氨是比水极性更强的质子碱,在水中存在OH-,NH4+和NH3\5H2O的电离平衡,因此氨水的加入会减少溶液中游离H+的浓度,从而具有抑制析氢的效果.但添加0.1 mol\5L-1和0.2 mol\5L-1 NH3\5H2O后的还原电流峰值无明显区别,推测是由于溶液中加入的氨过量导致.Gamali等[11]也发现,(NH4)2SO4溶液在在一定浓度范围内(0.125~2.6 mol\5L-1),析氢反应速率几乎与NH4+浓度无关,而同样条件下与Na2SO4相比,在电解液(NH4)2SO4中析氢反应的Tafel 斜率更大.他们认为在还原电位下,NH4+会还原产生NH3,而对氢的析出反应具有抑制作用,间接证实了本文的观点.Ramasamy等[12]在研究燃料电池中NH3\5H2O对不同金属表面析氢的催化作用时也得到了相似的结论:氨水的存在抑制了铂、镍等金属表面的析氢反应.
从图 3中可以看到,添加SeO2后,其析氢的峰电流从65 mA减少至38 mA,减小近50%,且析氢过电位增大,说明SeO2对氢的析出具有很显著的抑制作用;而添加(NH4)2SO3则不同,仅在电位为-1.3~-1.4 V区间表现出一定的抑制析氢作用,扫描电位继续负移,其峰值电流与未添加时基本一致.
2.3 添加剂对含Mn2+电解液析氢及锰沉积反应的影响进一步比较在 (NH4)2SO4电解质溶液中添加15 g\5L-1的Mn2+对锰沉积的影响以及锰沉积对析氢反应的影响,结果见图 4.添加Mn2+后,体系的还原电流大幅度减小,阴极峰值电流从80 mA降至53 mA,显然Mn2+的存在抑制了析氢反应.更为显著地是,负向扫描至-1.60V之前,其还原电流仅为未添加Mn2+体系的1/10,可见,尽管在此电位锰未开始沉积,但Mn2+的存在(可能是锰还原的某些前置还原步骤)对氢的析出表现出明显的抑制作用;继续负向扫描,随着锰沉积反应的进行,还原电流迅速增大.Xu等[13]的研究也证实Mn2+浓度越高时,还原反应中析氢反应所占比例越小.当电位正向扫描时,尽管电流大于负向扫描时的电流,但仍然远小于未添加Mn2+时的数值,同样表明锰沉积对析氢具有抑制作用.因此,从图 4可以看出,在没有辅助添加剂存在的情况下,锰可以沉积,但沉积效率较低,同时,锰的还原显著抑制析氢反应.

|
| 图 4 (NH4)2SO4电解液及(NH4)2SO4+Mn2+电解液中的CV曲线 Figure 4 CV curves in (NH4)2SO4 electrolyte and (NH4)2SO4+Mn2+ electrolyte,respectively |
在上述(NH4)2SO4+Mn2+电解质溶液中进一步添加0.1 mol\5L-1NH3\5H2O,考察锰沉积和析氢反应的变化,结果如图 5所示,阴极峰值电流进一步下降至0.025 A,不及(NH4)2SO4+Mn2+体系的1/2,进一步比较-1.40~-1.60 V间的还原电流,同样有所减小,显然,Mn2+和氨水共同存在时,其抑制析氢的作用进一步加强.正向扫描时,出现较大的氧化峰意味着添加氨水后体系的锰沉积量相比未添加的体系更大,而出现的两个氧化峰推测是Mn的氧化和H+还原相互竞争所致.结合图 2的结果表明,氨水在电解锰的阴极反应中同时具有抑制析氢和促进锰沉积的作用.

|
| 图 5 (NH4)2SO4+Mn2+电解液及添加NH3\5H2O 后体系的CV曲线 Figure 5 CV curves in (NH4)2SO4+Mn2+ electrolyte with and without NH3\5H2O,respectively |
进一步在上述体系中分别添加(NH4)2SO3和SeO2后,其CV曲线如图 6所示.可以看出,正向扫描的过程中未出现氧化峰,说明在(NH4)2SO3或者SeO2存在的体系中,Mn2+的沉积反应已经占主导作用.比较添加(NH4)2SO3或SeO2的差异可以看出,添加SeO2体系的阴极电流远小于添加(NH4)2SO3体系,表明SeO2抑制析氢的作用更为显著.分别积分上述两曲线的氧化电流和还原电流(还原电流代表锰沉积和析氢的总电流,氧化电流代表着沉积锰的氧化)可知,SeO2的氧化电量与还原电量的比值较大,意味着添加SeO2的体系具有更高的锰沉积效率.

|
| 图 6 (NH4)2SO4+Mn2+电解液中分别添加(NH4)2SO3及SeO2后体系的CV曲线 Figure 6 CV curves in (NH4)2SO4+Mn2+ electrolytes with (NH4)2SO3 or SeO2 as additive,respectively |
为了确认上述分析,同时比较了这两种添加剂对锰沉积的作用机理.称量不锈钢板沉积前后的质量,即可得到所得沉积锰的质量,根据(3)式[12]计算沉积过程的电流效率,结果列于表 1.
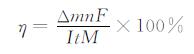
|
(3) |
| Additive | Δm/g | η/% |
| 0.03 g\5L-1SeO2 | 0.535 3 | 74.60 |
| 0.2 g\5L-1SO2 | 0.403 2 | 56.20 |
其中,Δm(g)表示沉积锰的质量,n为锰沉积反应电子数,F(C)为法拉第常数,I(A)为反应电流,t(s)为反应时间,M(g\5mol-1)为锰的摩尔质量.
对所得沉积层进行SEM和XRD测试,结果分别见图 7和图 8.

|
| 图 7 添加剂分别为0.2 g\5L-1 SO2(A)及0.03 g\5L-1SeO2(B)的电解锰片的SEM图 Figure 7 SEM images of manganese plates deposited with additives 0.2 g\5L-1 SO2(A) and 0.03g\5L-1 SeO2(B),respectively |

|
| 图 8 添加剂分别为0.2 g\5L-1 SO2及0.03g\5L-1SeO2的电解锰片XRD图 Figure 8 XRD spectra of manganese plates deposited with 0.2g\5L-1 SO2 or 0.03 g\5L-1 SeO2,respectively |
从表 1可以看出,沉积实验与CV曲线得到的结果一致,以SeO2为添加剂时锰的沉积效率更高.图 7的SEM图表明,两种添加剂条件下生成的锰团簇形状基本一致;但以SeO2为添加剂(图 7B)时,不同粒径的团簇交替堆积,表明沉积的锰更为致密,结合XRD标准图谱(JCPDS32-0637)和Dhananjayan[7]的研究可知,两种添加剂条件下所得到的锰均为具有立方结构的α-Mn,表明两种添加剂促进锰沉积的机理基本一致,由于SeO2具有更好的抑制析氢反应的作用,有利于锰紧密排列,而具有更高的沉积效率.
3 结 论本研究采用EQCM技术确认锰沉积和析氢反应的电势范围,采用传统三电极体系考察NH3\5H2O,(NH4)2SO3,SeO2等添加剂对锰沉积和析氢反应的影响.结果表明:
1) 在不锈钢阴极表面,氢的析出起始电位为-1.20 V,而锰沉积的起始电位为-1.60 V,证实析氢反应确实是难以彻底抑制的副反应;
2) 氨水和SeO2对析氢反应具有明显的抑制作用,而(NH4)2SO3对析氢反应的抑制作用并不显著;
3) 氨水对锰沉积具有一定的促进作用;
4) SeO2和(NH4)2SO3对锰的沉积均有一定的促进作用,且促进沉积的机理一致,由于SeO2具有更强的抑制析氢的作用,故具有更高的沉积效率.
| [1] | ALLMAND A J, CAMPBELL A N. The electrodeposition of manganese—Part Ⅱ[J]. Transactions of Faraday Society, 1924, 20(1) : 379–384. DOI:10.1039/TF9242000379 |
| [2] | LU J M, DREISINGER D, GLVCK T. Manganese electrodeposition—A literature review[J]. Hydrometallurgy, 2014, 141 : 105–116. DOI:10.1016/j.hydromet.2013.11.002 |
| [3] | ILEA P, POPESCU I C, URDA M, et al. The electrodeposition of manganese from aqueous solutions of MnSO4.IV:Electrowinning by galvanostatic electrolysis[J]. Hydrometallurgy, 1997, 46(1) : 149–156. DOI:10.1016/S0304-386X(97)00008-X |
| [4] | RADHAKRISHNAMURTHY P, REDDY A K N. The mechanism of manganese electrodeposition[J]. Journal of Applied Electrochemisty, 1974, 4 : 317–321. DOI:10.1007/BF00608973 |
| [5] | TILAK B, RAJAGOPALAN S R, REDD A K N. Buffer characteristics of manganous sulphate+ammonium sulphate electrodeposition baths[J]. Transactions of Faraday Society, 1962, 58 : 795–804. DOI:10.1039/TF9625800795 |
| [6] | GONG J, ZANGARI G. Electrodeposition and characterization of manganese coatings[J]. Journal of the Electrochemical Society, 2002, 149(4) : C209–C217. DOI:10.1149/1.1452117 |
| [7] | DHANANJAYAN N. Mechanism of electrodeposition of manganese in alpha and gamma modifications[J]. Journal of Electrochemistry Society, 1970, 117(8) : 1006–1011. DOI:10.1149/1.2407709 |
| [8] | RADHAKRISHNAMURTHY P, REDDY A K N. Mechanism of action of selenious acid in the electrodeposition of manganese[J]. Journal of Applied Electrochemisty, 1977, 7(2) : 113–117. DOI:10.1007/BF00611032 |
| [9] | NAYAK P K, MUNICHANDRAIAH N. An EQCM investigation of capacitance of MnO2 in electrolytes containing multivalent cations[J]. Journal of Electroanalytial Chemistry, 2012, 685 : 37–40. DOI:10.1016/j.jelechem.2012.09.003 |
| [10] | DIAZ-ARISTA P, ANTANO-LOPEZ R, MEAS Y, et al. EQCM study of the electrodeposition of manganese in the presence of ammonium thiocyanate in chloride-based acidic solutions[J]. Electrochimica Acta, 2006, 51(21) : 4393–4404. DOI:10.1016/j.electacta.2005.12.019 |
| [11] | GAMALI I V, STENDER V V. Hydrogen overvoltage on manganese[J]. Journal of Applied Chemistry of the USSR, 1962, 35(1) : 112–116. |
| [12] | RAMASAMY P, GERARDINE G B. Effect of ammonia on Pt, Ru, Rh, and Ni cathodes during the alkaline hydrogen evolution reaction[J]. Journal of Physical Chemistry C, 2013, 117(34) : 17429–17441. DOI:10.1021/jp405191c |
| [13] | XU F Y, DAN Z G, ZHAO W N, et al. Electrochemical analysis of manganese electrodeposition and hydrogen evolution from pure aqueous sulfate electrolytes with addition of SeO2[J]. Journal of Electroanalytical Chemistry, 2015, 741 : 149–156. DOI:10.1016/j.jelechem.2015.01.027 |