



2. Graduate School of Chinese Academy of Sciences, Beijing 100049, China;
3. ARC Centre of Excellence for Electromaterials Science(ACES), Intelligent Polymer Research Institute, AIIM Facility, Innovation Campus, University of Wollongong, NSW 2522, Australia;
4. Department of Biology, Indiana University Purdue University Indianapolis, Indianapolis, Indiana, USA
Objective: This study aimed to use a systematic approach to evaluate the current utilization, safety, and effectiveness of cell therapies for neurological diseases in human. And review the present regulations, considering United States (US) as a representative country, for cell transplantation in neurological disease and discuss the challenges facing the field of neurology in the coming decades. Methods: A detailed search was performed in systematic literature reviews of cellularbased therapies in neurological diseases, using PubMed, web of science, and clinical trials. Regulations of cell therapy products used for clinical trials were searched from the Food and Drug Administration (FDA) and the National Institutes of Health (NIH). Results: Seven most common types of cell therapies for neurological diseases have been reported to be relatively safe with varying degrees of neurological recovery. And a series of regulations in US for cellular therapy was summarized including preclinical evaluations, sourcing material, stem cell manufacturing and characterization, cell therapy product, and clinical trials. Conclusions: Stem cell-based therapy holds great promise for a cure of such diseases and will value a growing population of patients. However, regulatory permitting activity of the US in the sphere of stem cells, technologies of regenerative medicine and substitutive cell therapy are selective, theoretical and does not fit the existing norm and rules. Compiled well-defined regulations to guide the application of stem cell products for clinical trials should be formulated.
Stem cells can self‐renew,proliferate,and differentiate into specialized cells for cell replacement therapy of a wide variety of diseases,including neurological disorders. In a mouse model of PD,grafts of neural stem cells (NSC) survive in the midbrains and significantly improve behavior performance[1]. The human counterparts of such stem cells are also functional in pre‐clinical investigations[2]. Because of a lack of quality control for such type of cell products,in August of 2010,the Food and Drug Administration (FDA) approved the first proposed clinical trials of human embryonic stem cells (hESC)‐derived oligodendrocyte progenitor cells. As the first product of hESC derivatives approved by the FDA for clinical trials, GRNOPC1 were primarily examined for the treatment of SCI[3]. Other clinical trials were approved shortly after to evaluate the use of hESC‐derived retinal pigment epithelium in age‐related macular degeneration by ACT,Inc.,and NSC in treating Pelizaeus‐ Merzbacher disease by SCI,Inc.[4] The past 10 years has witnessed an increase in the number of clinical trials involving stem cell products in United States (US),as shown in Table 1. Although not all trials will generate positive results for a specific entity of product, these Phase Ⅰ or Ⅱ trials will accumulate valuable data that will be informative for future stem cell‐based therapy[5]. Here,we summarize recent advances in cell transplantation for ND and discuss the policies and regulations of cell therapy in the United States (US).
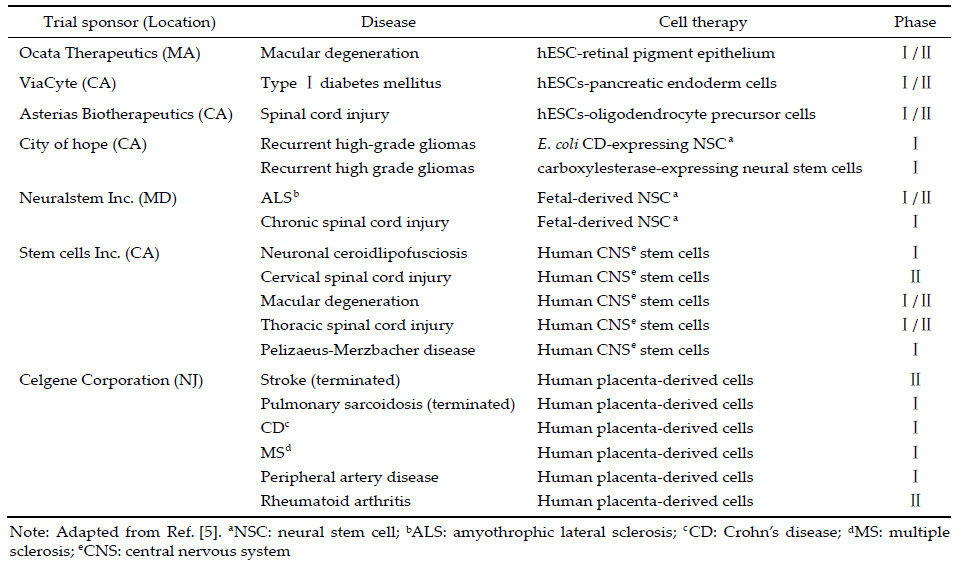
In August 2010,as the first product of hESC derivatives,hESC‐derived oligodendrocyte progenitor cells were approved by the FDA for clinical trial, and GRNOPC1 was to be used for the treatment of SCI[3]. However,in the preclinical study,cystogenesis occurred when cells were transplanted into animals, and the clinical trial was postponed for two years until this issue was resolved. Subsequently,other clinical trials ofhESC‐NSC in treating Pelizaeus‐Merzbacher disease were conducted by SCI[4],and the specific treatment effect is being examined.
3 Preclinical evaluations of neural cells differentiated from human stem cells In order to cure ND using hESC‐derived neural cells, a growing number of preclinical studies have been carried out in animal models varying from rodents to primates. The types of cells used in these studies include neural progenitors and terminal differentiated neural cells. As described above,MSC can be differentiated into neural‐like cells[42, 43] and have shown positive effects when transplanted into animal models of ND[44].Neural cells derived from other types of pluripotent stem cells as well were proved functional in preclinical studies. For instance,glial‐rich neural progenitors derived from human iPSC were transplanted into rodent models of mutant superoxide dismutase 1 (SOD1)‐mediated ALS[45]. These transplanted cells then converted into astrocytes and expanded the lifespan of recipient mice,indicating the efficacy of human iPSC‐derived glial progenitors for cell therapy of ALS. In animal models of SCI,human glial restricted progenitors (hGRP) and hGRP‐derived astrocytes survived following engraftment into the injured spinal cord,migrated extensively in the rostral and caudal directions into the adjacent white matter,and expressed a variety of phenotypic markers including GFAP[46]. This research also showed that embryonic astrocytes alone were not sufficient to promote functional recovery through long‐distance axonal regeneration,suggesting a necessity of combining cell transplantation and other therapeutic interventions such as neurotrophins[47],cAMP[48] and chondroitinase[49]. Nevertheless,because of the high heterogeneity of glia,it should be noted that not all astroglia have the same function in curing ND. Both hESC‐derived Olig2+ and Olig2‐ progenitors could be differentiated into astroglia,respectively. However, upon transplanted into rat model of ischemic stroke, Olig2+/‐ astroglia exhibit distinctions from the Olig2‐ and the mixed population of astroglia and significantly improved learning and memory of the ischemic stroke rats[50].
4 Regulatory agencies of stem cells and translational medicine in America In the 2000s,the United States (US) informed the world that work with stem cells is immoral and should be stopped. The stage of stem cell research is rudderless, and this greatly slowed the research on stem cells. Finally,the ban on using federal funding for human embryonic stem cell research was lifted on March 9, 2009,when Barack Hussein Obama Jr.,the new President,issued executive order 13505. A door was re‐opened for human embryonic stem cell research and regenerative medicine in the US triggering the advance of reliable scientific research involving human stem cells and the approval of the first clinical study of hESC by the FDA[51]. In the same year,an extensive and detailed guideline: “National Institutes of Health Guidelines for Human Stem Cell Research” (Guidelines) was finalized by the National Institutes of Health (NIH)[52]. Thereafter,with the advance of stem cell research and clinical translation,a series of customized guidelines,regulations,and laws were mandated in the following years. For example,two were published for cellular therapy for cardiac disease and for allogeneic pancreatic islet cell products (Cellular Therapy for Cardiac Disease,Considerations for Allogeneic Pancreatic Islet Cell Products). These documents emphasize that any processing and manufacture of cellular products should be conducted with stringent,independent review and supervision to ensure the safety,integrity, and efficacy for use in patients. 4.1 Regulations on sourcing material hESC lines are derived from pre‐implanted embryos; this is fraught with disputes regarding the onset of human personhood and reproduction. hESC researches are surrounded by ethical and political controversy (Table 2). According to the guidelines,the current regulatory structure of the Institutional Review Board (IRB) review under the common rule (45 CFR Part 46, Subpart A) addresses the core ethical principles needed for appropriate oversight of hESC derivation. Several other methods of deriving stem cells have fewer ethical concerns,for example,the reprogramming of somatic cells to produce iPSC avoids the ethical problems specific to hESC. In addition,iPSC used for the original donor can go through a minimum screening and has less immunogenicity compared with hESC. Nevertheless,iPSC have not been used in clinical trial in the US iPSC and their derivatives should be subjected to additional preclinical tests because of the potential risks of genomic instability caused by reprogramming or the possible functional defects owing to the nature of unnatural entities.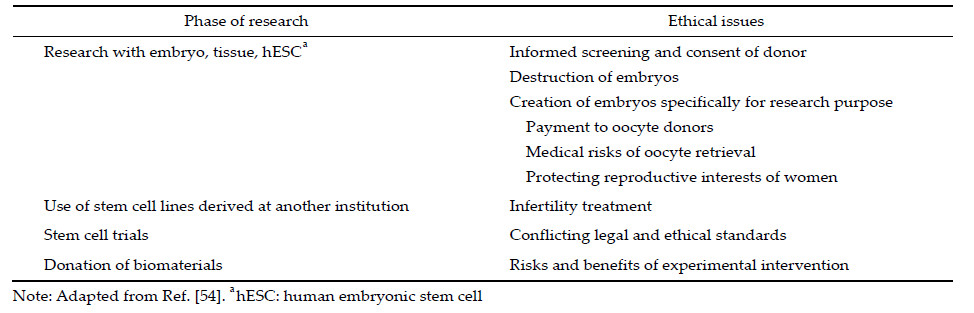
The only way to establish eligible hESC lines can be registered on NIH (http://grants.nih.gov/stem_cells/ registry/current.htm) to avoid burdensome and repetitive assurances from multiple funding applicants, or to demonstrate compliance with the specific pro‐ cedural requirements of guidelines by submitting an assurance with supporting information for administrative review by the NIH. In President Bush’s era, only certain existing embryonic stem cell lines were recognized by the NIH. Currently,increasing number of new hESC lines are eligible for NIH funding,and federal funding is also available to carry out research with hESC lines not on the NIH list and to derive new hESC lines from frozen embryos donated for research if a woman who received in vitro fertilization (IVF) has determined they are no longer needed for reproductive purposes. However,derivation of stem cells using somatic cell nuclear transfer is still not permitted under federal funding[53, 54].
Currently,cells used in studies from a wide variety of sources including autologous or allogeneic,undifferentiated stem/progenitor cells,or terminally differentiated cells have been submitted to the FDA for review. To ensure safety of the manufacturing process of cells,several aspects need to be considered, including the eligibility of donors selected to provide the source material through screening and testing. Moreover,the information of the donor should be a specific conservation (Informed Consent/Institutional Review Boards: 21 CFR Parts 50/56). In detail,(1) the donor will be screened for infectious and possibly genetic diseases. Pre‐implantation genetic diagnosis (PGD) of the donation of embryos is allowed by NIH, although this may result in the identification of chromosomal abnormalities that would make the embryos medically unsuitable for clinical use; (2) the donated cells may be subject to genetic modification by the investigator; (3) disclosure of medical and other relevant information that will be retained,and the specific steps that will be taken to protect donor privacy and confidentiality of retained information, including the date at which donor information will be destroyed,if applicable; (4) the donor should be informed that in the case of pluripotent stem cells,the ability to destroy all samples may be limited and that with newer genetic techniques,complete anonymity may not be feasible; (5) disclosure that any resulting cells,lines,or other stem cell‐derived products may have commercial potential,and whether any commercial and intellectual property rights will reside with the institution conducting the research. The FDA may be agnostic about the source of stem cell,apart from assurance of product characteristics. The investigator and research institution do not have access to identifiable private information related to the cell line, and a written agreement is obtained from the holder of the identifiable private information related to the cell line provided that such information will not be released to the investigator under any circumstances (45 CFR Part 46). In this case,the research may be considered to not involve human subjects.
4.2 Regulations for stem cell manufacturing and characterization As for the production process,a detailed description of all programs of standard operating procedures (SOPs) must be given related to product quality as a traditional medicinal product. The procurement of tissue from a human donor may not fall under the jurisdiction of current Good Manufacturing Practice (cGMP,21 CFR 211),but should be conducted using Good Laboratory practices (GLP,21 CFR Part 58). Various types of reagents may be used to manufacture the cellular component of a product. These include reagents that promote cellular replication and induce differentiation,used to select targeted cell populations, specifically,serum,peptides,cytokines,and monoclonal antibodies,and culture medium. The reagents must be duly qualified. Demonstration of manufacturing controls was evidenced by strict adherence to SOPs and quality control assessment of manufacturing intermediates and testing of the final cellular preparation.Criteria for release of cells for transfer to research subjects in trials must minimize risk from cultureacquired abnormalities. It should also follow regulatory guidelines including universal precautions to minimize the risks of contamination,infection,and pathogen transmission. One of the crucial requirements to enable clinical use of these cells and embryo is to eliminate the risk of xeno‐transmitted infections and immunoreactions caused by animal products. This would enable better expansion of cells in xeno culture system,including medium,digestive tool, and cryoprotectant,prior to transplant. The FDA explains a testing approach for characterization of cells. This involves development of quantitative analytical techniques for conducting a panel of tests including morphology evaluation,detection of phenotype‐specific cell surface antigens,genomic/proteomic analysis, and assessment of unique biochemical markers to determine the characterization of stem cells.
4.3 Regulations for preclinical cell therapy products (CTPs) For the overall consideration of the design of preclinical studies to support early‐phase clinical trials, the FDA has published guidance entitled “Guidance for Industry: Preclinical Assessment of Investigational Cellular and Gene Therapy Products”. All preclinical studies testing the safety and efficacy should be designed in a way that supports a precise,accurate, and unbiased measure of clinical promise. CTPs vary with respect to characteristics such as the formulation including combination with a scaffold or other non‐cellular component,genetic relationship of the cells to the patient (autologous,allogeneic,xenogeneic), and cell source. CTPs can be generally classified as stem cell‐derived CTPs and mature/functionally differentiated cell‐derived CTPs. CTPs must be in compliance with cGMP,including quality control and quality assurance programs during translation from “bench to bedside”,to minimize the risks of contamination, infection,and pathogen transmission.To determine whether it is reasonable to provide granting permissions for clinical trials,the FDA evaluates the potential risk based on results derived from the analytical assessment of CTP characteristics and safety testing. Preclinical in vitro and in vivo proof‐of‐concept,pharmacology,and toxicology studies are conducted to establish feasibility and rationale for clinical use. These studies also provide the scientific basis to support the conclusion that it is reasonably safe to conduct the proposed clinical investigations (21 CFR 312.23(a) (8)). Preclinical in vitro animal model studies are the principal means of testing. Various models that have been considered include immunosuppressive agents in immune‐competent animals; genetically immunodeficient animals; humanized animals; administration into an immuneprivileged site; or a combination of these scenarios (PHS Guideline on Infectious Disease Issues in Xenotransplantation; January 2001). Important design elements for preclinical animal studies,include (1) selection of relevant disease/injury models; (2) testing of product intended for clinical administration; (3) using route and method of delivery comparable to clinical plan; (4) optimal timing of intervention relative to disease/ injury onset; (5) duration necessary to assess potential adverse safety events and durable biological activity; (6) need for immunocompetence modification of animal model[55]. In addition to the plasticity and unlimited capacity of stem cells,ES cells can themselves form teratomas. Overall,CTPs to be used in clinical trials must first be rigorously characterized to assess potential toxicities through in vitro studies and in animal studies. In some cases,the donor may receive a treatment prior to the harvest of the source material. If the donor is also the trial subject,such pretreatment may add to the overall risk to the subject. Similarly, some CTPs require pretreatment of the recipient, such as with immune modification or myeloablative conditioning to facilitate cell survival. In such cases, the risks associated with the pre‐treatment should be considered in the overall benefit‐risk assessment.
4.4 Regulations for clinical trial In the United States,all clinical research involving cells or test articles regulated as drugs,devices,and biological products are subject to the FDA regulations governing investigational new drugs (INDs) or devices (IDEs) (21 CFR Parts 312 or 812),biologics regulations (21 CFR 600),and cGMP. In particular,the US federal regulations divide cellular therapy into two sections of the Public Health Service Act (PHSA),referred as “361 produces” and “351 produces”. Traditional blood, bone marrow progenitor cells,and other tissues for transplantation are classified into section 361 produces. The FDA regulates that cells or tissues used for therapeutic purposes and the regulation that pertains to process of 361 produces are codified under the Good Tissue Practice (GTP)[56]. Part of CTPs that is only used for safety and effectiveness assessment, can be pertained to process of 351 produces,the requirements of which are not as strict as those for 361 produces.For early phase clinical trials,especially first‐inhuman trials,the regulations in 21 CFR Part 312 emphasize the importance of the assessment of trial risks and the safeguards for trial subjects. Regulations on cells,tissue,and cellular and tissue‐based products (HCT/Ps) are ruled in CFR 21,part 1271. Moreover, guidance: “Considerations for the Design of Early‐Phase Clinical Trials of Cellular and Gene Therapy Products” published by the FDA is the explicit guide for clinical trial design of CTPs. The objectives of early‐phase clinical include (1) dose exploration: initial tests of a novel product should be tested under low risk condition of low dose before escalating to a higher dose to determine the relationship with potential adverse reactions,and the best route of administration; (2) feasibility assessments: CTPs sometimes require specialized equipment or novel procedures for customized preparation of products,special handling of products administration,or adjunctive therapy. In these cases,sponsors should consider designing early phase trials to identify and characterize any technical or logistic issues with manufacturing and administering the product; (3) activity assessments: a common secondary objective of early phase trials is to obtain preliminary assessments of product activity,and such trials can use both short‐term responses and longterm outcome. It could suggest the potential efficacy of the products. Such proof‐of‐concept data can support subsequent clinical development. For CTPs,activity assessments might include specialized measures such as gene expression,cell engrafting,or morphologic alterations,and more common measures such as changes in immune function,tumor shrinkage,or physiologic responses of various types. In addition, choice of study population is stringent. The potential for benefit might depend on the choice of study population,including healthy volunteers,disease stage,or severity of patients or other special group such as pediatrics. For CTPs,the potential instability of cells deemed the trials unacceptable for healthy volunteers. Selection of sick volunteers involves consideration of the potential benefits and the ability of the study population to provide interpretable data. For the principle of scientific necessity,children should not be enrolled in a clinical investigation unless necessary to answer an important scientific question about the health and welfare of children (21 CFR 56.111 (a)(1) and (b)).
Late phase trials are aimed at providing decisive evidence of clinical utility. Currently,most of CTPs are tested in the early phase,and very few in phase Ⅲ. Late phase trials benefit by the use of clinical measures and response by monitoring over a longer,more clinically relevant period. To preserve the ability to draw valid conclusions about the clinical benefit of late phase clinical trials,use of generally randomized and comparator arms to avoid artificial factor makes the trials more reasonable,objective,and true. Moreover, careful consideration of appropriate product features, proof‐of‐concept testing,and the design of clinical trials will enable balanced case‐by‐case consideration of proposed therapeutic trials[57].
5 Conclusion and outlook Thus far,the policy of the US seems obscure and unclear. The regulatory permitting activities of the US in the sphere of stem cells,technologies of regenerative medicine,and substitutive cell therapy are selective,theoretical,and do not fit the existing norm and rules,for example,set for the pharmaceuticals. More clear and detailed policy should be formulated. Currently,detailed policies have been formulated for cardiac disease (Guidance for Industry: Cellular therapy for Cardiac Disease) and diabetes (Considerations for Allogeneic Pancreatic Islet Cell Products). However,there is no tailor‐made guidance for cell therapies of ND thus far.As a whole,the field of cell transplantation is advancing swiftly with the prosperous banking and manufacturing of stem cells,which will lead to effective therapies for patients. In a framework that facilitates the healthy translation of stem cell research for ND,clinical trials with stem cell products should consider scientific evidence and strictly follow the FDA regulatory guidelines to ensure safety and efficacy. More reliable approaches for precise evaluation of safety and efficacy should be developed so that the most valuable information could be collected for prompt assessment of innovative cell therapy for disorders such as ND.
The authors have no financial interest to disclose regarding the article.
| [1] | Kim JH, Auerbach JM, Rodríguez-Gómez JA, Velasco I, Gavin D, Lumelsky N, Lee SH, Nguyen J, Sánchez-Pernaute R, Bankiewicz K, McKay R. Dopamine neurons derived from embryonic stem cells function in an animal model of Parkinson's disease. Nature 2002, 418(6893):50-56. |
| Click to display the text | |
| [2] | Muotri AR, Nakashima K, Toni N, Sandler VM, Gage FH. Development of functional human embryonic stem cellderived neurons in mouse brain. Proc Natl Acad Sci USA 2005, 102(51):18644-18648. |
| Click to display the text | |
| [3] | Lebkowski J. GRNOPC1:the world's first embryonic stem cell-derived therapy. Interview with Jane Lebkowski. Regener Med 2011, 6(6 Suppl):11-13. |
| Click to display the text | |
| [4] | Schwartz SD, Hubschman J-P, Heilwell G, Franco-Cardenas V, Pan CK, Ostrick RM, Mickunas E, Gay R, Klimanskaya I, Lanza R. Embryonic stem cell trials for macular degeneration:A preliminary report. Lancet 2012, 379(9817):713-720. |
| Click to display the text | |
| [5] | Trounson A, McDonald C. Stem cell therapies in clinical trials:Progress and challenges. Cell Stem Cell 2015, 17(1):11-22. |
| Click to display the text | |
| [6] | Corrigan JD, Selassie AW, Orman JA. The epidemiology of traumatic brain injury. J Head Trauma Rehabil 2010, 25(2):72-80. |
| Click to display the text | |
| [7] | Mayeux R. Epidemiology of neurodegeneration. Ann Rev Neurosci 2003, 26:81-104. |
| Click to display the text | |
| [8] | Bianchi G, Muraglia A, Daga A, Corte G, Cancedda R, Quarto R. Microenvironment and stem properties of bone marrow-derived mesenchymal cells. Wound Repair Regen 2001, 9(6):460-466. |
| Click to display the text | |
| [9] | Filioli Uranio M, Valentini L, Lange-Consiglio A, Caira M, Guaricci AC, L'Abbate A, Catacchio CR, Ventura M, Cremonesi F, Dell'Aquila ME. Isolation, proliferation, cytogenetic, and molecular characterization and in vitro differentiation potency of canine stem cells from foetal adnexa:A comparative study of amniotic fluid, amnion, and umbilical cord matrix. Mol Reprod Dev 2011, 78(5):361-373. |
| Click to display the text | |
| [10] | Xiao L, Tsutsui T. Characterization of human dental pulp cells-derived spheroids in serum-free medium:Stem cells in the core. J Cell Biochem 2013, 114(11):2624-2636. |
| Click to display the text | |
| [11] | Jones BJ, McTaggart SJ. Immunosuppression by mesenchymal stromal cells:From culture to clinic. Exp Hematol 2008, 36(6):733-741. |
| Click to display the text | |
| [12] | Kisiel AH, McDuffee LA, Masaoud E, Bailey TR, Esparza Gonzalez BP, Nino-Fong R. Isolation, characterization, and in vitro proliferation of canine mesenchymal stem cells derived from bone marrow, adipose tissue, muscle, and periosteum. Am J Vet Res 2012, 73(8):1305-1317. |
| Click to display the text | |
| [13] | Bossolasco P, Cova L, Calzarossa C, Rimoldi SG, Borsotti C, Deliliers GL, Silani V, Soligo D, Polli E. Neuro-glial differentiation of human bone marrow stem cells in vitro. Exp Neurol 2005, 193(2):312-325. |
| Click to display the text | |
| [14] | Levy YS, Bahat-Stroomza M, Barzilay R, Burshtein A, Bulvik S, Barhum Y, Panet H, Melamed E, Offen D. Regenerative effect of neural-induced human mesenchymal stromal cells in rat models of Parkinson's disease. Cytotherapy 2008, 10(4):340-352. |
| Click to display the text | |
| [15] | Woodbury D, Schwarz EJ, Prockop DJ, Black IB. Adult rat and human bone marrow stromal cells differentiate into neurons. J Neurosci Res 2000, 61(4):364-370. |
| Click to display the text | |
| [16] | Karussis D, Karageorgiou C, Vaknin-Dembinsky A, Gowda-Kurkalli B, Gomori JM, Kassis I, Bulte JWM, Petrou P, Ben-Hur T, Abramsky O, Slavin S. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch Neurol 2010, 67(10):1187-1194. |
| Click to display the text | |
| [17] | Bang OY, Lee JS, Lee PH, Lee G. Autologous mesenchymal stem cell transplantation in stroke patients. Ann Neurol 2005, 57(6):874-882. |
| Click to display the text | |
| [18] | Wang S, Cheng HB, Dai GH, Wang XD, Hua RR, Liu XB, Wang PS, Chen GM, Yue W, An YH. Umbilical cord mesenchymal stem cell transplantation significantly improves neurological function in patients with sequelae of traumatic brain injury. Brain Res 2013, 1532:76-84. |
| Click to display the text | |
| [19] | Westwood CC, Clements MO. The biology of human mesenchymal stem cells. In Stem Cell Repair and Regeneration. Levicar N, Habib NA, Gordon MY, Dimarakis I, Eds. London, UK:Imperial College Press, 2008, Vol. 3, pp1-20. |
| Click to display the text | |
| [20] | Bryukhovetskiy AS, Bryukhovetskiy IS. Effectiveness of repeated transplantations of hematopoietic stem cells in spinal cord injury. World J Transplant 2015, 5(3):110-128. |
| Click to display the text | |
| [21] | Mancardi GL, Sormani MP, Gualandi F, Saiz A, Carreras E, Merelli E, Donelli A, Lugaresi A, Di Bartolomeo P, Rottoli MR, Rambaldi A, Amato MP, Massacesi L, Di Gioia M, Vuolo L, Curro D, Roccatagliata L, Filippi M, Aguglia U, Iacopino P, Farge D, Saccardi R. Autologous hematopoietic stem cell transplantation in multiple sclerosis:A phase Ⅱ trial. Neurology 2015, 84(10):981-988. |
| Click to display the text | |
| [22] | Chen JL, Sanberg PR, Li Y, Wang L, Lu M, Willing AE, Sanchez-Ramos J, Chopp M. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke 2001, 32(11):2682-2688. |
| Click to display the text | |
| [23] | Xia GZ, Hong XR, Chen XM, Lan FH, Zhang GC, Liao LM. Intracerebral transplantation of mesenchymal stem cells derived from human umbilical cord blood alleviates hypoxic ischemic brain injury in rat neonates. J Perinat Med 2010, 38(2):215-221. |
| Click to display the text | |
| [24] | Kim ES, Ahn SY, Im GH, Sung DK, Park YR, Choi SH, Choi SJ, Chang YS, Oh W, Lee JH, Park WS. Human umbilical cord blood-derived mesenchymal stem cell transplantation attenuates severe brain injury by permanent middle cerebral artery occlusion in newborn rats. Pediatr Res 2012, 72(3):277-284. |
| Click to display the text | |
| [25] | Newcomb JD, Sanberg PR, Klasko SK, Willing AE. Umbilical cord blood research:Current and future perspectives. Cell Transplant 2007, 16(2):151-158. |
| Click to display the text | |
| [26] | Taguchi A, Soma T, Tanaka H, Kanda T, Nishimura H, Yoshikawa H, Tsukamoto Y, Iso H, Fujimori Y, Stern DM, Naritomi H, Matsuyama T. Administration of CD34+ cells after stroke enhances neurogenesis via angiogenesis in a mouse model. J Clin Invest 2004, 114(3):330-338. |
| Click to display the text | |
| [27] | Sanchez-Ramos JR, Song SJ, Kamath SG, Zigova T, Willing A, Cardozo-Pelaez F, Stedeford T, Chopp M, Sanberg PR. Expression of neural markers in human umbilical cord blood. Exp Neurol 2001, 171(1):109-115. |
| Click to display the text | |
| [28] | Beukelaers P, Vandenbosch R, Caron N, Nguyen L, Moonen G, Malgrange B. Cycling or not cycling:Cell cycle regulatory molecules and adult neurogenesis. Cell Mol Life Sci 2012, 69(9):1493-1503. |
| Click to display the text | |
| [29] | Masiulis I, Yun S, Eisch AJ. The interesting interplay between interneurons and adult hippocampal neurogenesis. Mol Neurobiol 2011, 44(3):287-302. |
| Click to display the text | |
| [30] | Gould E, Reeves AJ, Graziano MSA, Gross CG. Neurogenesis in the neocortex of adult primates. Science 1999, 286(5439):548-552. |
| Click to display the text | |
| [31] | Eriksson PS, Perfilieva E, Björk-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH. Neurogenesis in the adult human hippocampus. Nat Med 1998, 4(11):1313-1317. |
| Click to display the text | |
| [32] | Sánchez-Pernaute R, Studer L, Ferrari D, Perrier A, Lee H, Viñuela A, Isacson O. Long-term survival of dopamine neurons derived from parthenogenetic primate embryonic stem cells (cyno-1) after transplantation. Stem Cells 2005, 23(7):914-922. |
| Click to display the text | |
| [33] | Sanai N, Tramontin AD, Quiñones-Hinojosa A, Barbaro NM, Gupta N, Kunwar S, Lawton MT, McDermott MW, Parsa AT, Manuel-García Verdugo J, Berger MS, Alvarez-Buylla A. Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature 2004, 427(6976):740-744. |
| Click to display the text | |
| [34] | Emborg ME, Ebert AD, Moirano J, Peng S, Suzuki M, Capowski E, Joers V, Roitberg BZ, Aebischer P, Svendsen CN. GDNF-secreting human neural progenitor cells increase tyrosine hydroxylase and VMAT2 expression in MPTPtreated cynomolgus monkeys. Cell Transplant 2008, 17(4):383-395. |
| Click to display the text | |
| [35] | Lima C, Pratas-Vital J, Escada P, Hasse-Ferreira A, Capucho C, Peduzzi JD. Olfactory mucosa autografts in human spinal cord injury:A pilot clinical study. JSpinal Cord Med 2006, 29(3):191-203. |
| Click to display the text | |
| [36] | Wu J, Sun T, Ye C, Yao J, Zhu B, He H. Clinical observation of fetal olfactory ensheathing glia transplantation (OEGT) in patients with complete chronic spinal cord injury. Cell Transplant 2012, 21(Suppl 1):S33-S37. |
| Click to display the text | |
| [37] | Granger N, Blamires H, Franklin RJM, Jeffery ND. Autologous olfactory mucosal cell transplants in clinical spinal cord injury:A randomized double-blinded trial in a canine translational model. Brain 2012, 135:3227-3237. |
| Click to display the text | |
| [38] | Tabakow P, Jarmundowicz W, Czapiga B, Fortuna W, Miedzybrodzki R, Czyz M, Huber J, Szarek D, Okurowski S, Szewczyk P, Gorski A, Raisman G. Transplantation of autologous olfactory ensheathing cells in complete human spinal cord injury. Cell Transplant 2013, 22(9):1591-1612. |
| Click to display the text | |
| [39] | Chen L, Huang HY, Xi HT, Xie ZH, Liu RW, Jiang Z, Zhang F, Liu YC, Chen D, Wang QM, Wang HM, Ren YS, Zhou CM. Intracranial transplant of olfactory ensheathing cells in children and adolescents with cerebral palsy:A randomized controlled clinical trial. Cell Transplant2010, 19(2):185-191. |
| Click to display the text | |
| [40] | Piepers S, van den Berg LH. No benefits from experimental treatment with olfactory ensheathing cells in patients with ALS. Amyotroph Lateral Scler 2010, 11(3):328-330. |
| Click to display the text | |
| [41] | Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282(5391):1145-1147. |
| Click to display the text | |
| [42] | Barzilay R, Melamed E, Offen D. Introducing transcription factors to multipotent mesenchymal stem cells:Making transdifferentiation possible. Stem Cells 2009, 27(10):2509-2515. |
| Click to display the text | |
| [43] | Tropel P, Platet N, Platel JC, Noël D, Albrieux M, Benabid AL, Berger F. Functional neuronal differentiation of bone marrow-derived mesenchymal stem cells. Stem Cells 2006, 24(12):2868-2876. |
| Click to display the text | |
| [44] | Liu Z, He B, Zhang RY, Zhang K, Ding Y, Ruan JW, Ling EA, Wu JL, Zeng YS. Electroacupuncture promotes the differentiation of transplanted bone marrow mesenchymal stem cells preinduced with neurotrophin-3 and retinoic acid into oligodendrocyte-like cells in demyelinated spinal cord of rats. Cell Transplant 2015, 24(7):1265-1281. |
| Click to display the text | |
| [45] | Haas C, Fischer I. Human astrocytes derived from glial restricted progenitors support regeneration of the injured spinal cord. J Neurotraum 2013, 30(12):1035-1052. |
| Click to display the text | |
| [46] | Cao QL, Xu XM, Devries WH, Enzmann GU, Ping PP, Tsoulfas P, Wood PM, Bunge MB, Whittemore SR. Functional recovery in traumatic spinal cord injury after transplantation of multineurotrophin-expressing glial-restricted precursor cells. J Neurosci 2005, 25(30):6947-6957. |
| Click to display the text | |
| [47] | Nout YS, Culp E, Schmidt MH, Tovar CA, Pröschel C, Mayer-Pröschel M, Noble MD, Beattie MS, Bresnahan JC. Glial restricted precursor cell transplant with cyclic adenosine monophosphate improved some autonomic functions but resulted in a reduced graft size after spinal cord contusion injury in rats. Exp Neurol 2011, 227(1):159-171. |
| Click to display the text | |
| [48] | Filous AR, Miller JH, Coulson-Thomas YM, Horn KP, Alilain WJ, Silver J. Immature astrocytes promote CNS axonal regeneration when combined with chondroitinase ABC. Dev Neurobiol 2010, 70(12):826-841. |
| Click to display the text | |
| [49] | Kondo T, Funayama M, Tsukita K, Hotta A, Yasuda A, Nori S, Kaneko S, Nakamura M, Takahashi R, Okano H, Yamanaka S, Inoue H. Focal transplantation of human iPSC-derived glial-rich neural progenitors improves lifespan of ALS mice. Stem Cell Reports 2014, 3(2):242-249. |
| Click to display the text | |
| [50] | Jiang P, Chen C, Wang RM, Chechneva OV, Chung SH, Rao MS, Pleasure DE, Liu Y, Zhang QG, Deng WB. hESCderived Olig2+ progenitors generate a subtype of astroglia with protective effects against ischaemic brain injury. Nat Commun 2013, 4:2196. |
| Click to display the text | |
| [51] | Strauss S. Geron trial resumes, but standards for stem cell trials remain elusive. Nat Biotechnol 2010, 28(10):989-990. |
| Click to display the text | |
| [52] | National Institutes of Health Guidelines on Human Stem Cell Research. 2009, Available from:http://stemcells.nih.gov/policy/pages/2009guidelines.aspx. |
| Click to display the text | |
| [53] | Dawson L, Bateman-House AS, Mueller AD, Bok H, Brock DW, Chakravarti A, Greene M, King PA, O'Brien SJ, Sachs DH, Schill KE, Siegel A, Solter D, Suter SM, Verfaillie CM, Walters LB, Gearhart JD, Faden RR. Safety issues in cellbased intervention trials. Fertil Steril 2003, 80(5):1077-1085. |
| Click to display the text | |
| [54] | Lo B, Parham L. Ethical issues in stem cell research. Endocr Rev 2009, 30(3):204-213. |
| Click to display the text | |
| [55] | Fink DW Jr. FDA regulation of stem cell-based products. Science 2009, 324(5935):1662-1663. |
| Click to display the text | |
| [56] | Astori G, Soncin S, Lo Cicero V, Siclari F, Sürder D, Turchetto L, Soldati G, Moccetti T. Bone marrow derived stem cells in regenerative medicine as advanced therapy medicinal products. Am J Transl Res 2010, 2(3):285-295. |
| Click to display the text | |
| [57] | Moos M Jr. Stem-cell-derived products:An FDA update. Trends Pharmacol Sci 2008, 29(12):591-593. |
| Click to display the text |