

硝基呋喃类药物是人工合成的具有5-硝基结构的广谱抗菌抗生素,常见的呋喃唑酮(FZD)、呋喃它酮(FTD)、呋喃西林(NFZ)和呋喃妥因(NFT)四种药物,其对应的代谢物分别为3-氨基-2-噁唑烷基酮(AOZ)、5-甲基-吗啉-3-氨基-2-噁唑烷基酮(AMOZ)、氨基脲(SEM)和1-氨基-2-内酰脲(AHD)。硝基呋喃类药物曾作为治疗和预防埃希氏菌和沙门氏菌引起的哺乳动物消化道疾病的药物和饲料添加剂,但是由于硝基呋喃类药物对人类健康具有潜在危害,欧盟2377/90/EEC 号令已全面禁止使用呋喃类抗菌药物,要求以代谢物为目标分析物,检测硝基呋喃原药残留物,并要求方法灵敏度达到1μg/kg。中国、美国、日本也相继禁止在食用动物中使用呋喃类药物作为生长剂和杀菌剂,方法检出限要求达到0.5μg/kg[1]。硝基呋喃类药物成为各个国家限制第三国出口的贸易壁垒以及保障国民健康的安全渠道,因此开展该项目的检测具有非常重要的意义。
目前针对水产品中硝基呋喃类药物的检测主要分为液相色谱法(LC)和液相质谱法(LC/MS/MS),其中采用高效液相色谱法能够同时测定呋喃唑酮、呋喃西林、呋喃妥因和呋喃它酮药物残留量[2-3],但是方法由于检出限较高,干扰物质难以区分受到局限。相比较 LC/MS/MS 检出限低,抗干扰能力强,在硝基呋喃类药物检测中具有非常大的优势,所以受到广泛的应用[4-6]。国家标准《动物源性食品中硝基呋喃类药物代谢物残留量检测方法高效液相色谱串联质谱法》(GB/T21311-2007)[7]中推荐使用 LC/MS/MS 法测定硝基呋喃类药物。标准中详细介绍使用三重四极杆类液相色谱质谱联用仪分析样品中硝基呋喃类代谢物的方法,但是使用该方法检测硝基呋喃存在假阳性,尤其是呋喃唑酮、呋喃它酮,由于基质干扰而无法准确检测化合物,所以有待进一步改进。
由于水产品中内源性物质的存在,即使使用LC/MS/MS,当保留时间,离子对比例与目标化合物相似的情况下,多反应监控(MRM)扫描模式难以确证出峰化合物真伪,导致呋喃唑酮、呋喃它酮检测结果出现偏差。当样品中目标化合物含量较高,可采用三重四极杆的子离子扫描模式采集化合物的二级子离子,与标准品子离子进行匹配从而实现定性检测,当化合物含量不高,传统三重四极杆难以采集有效的二级子离子,进而无法实现定性功能,尤其在低浓度或检出限附近时,对化合物的确证是极大的挑战。为了能够准确检测水产品中呋喃唑酮、呋喃它酮的含量,排除假阳性可能,实验中采用 QTRAP 类质谱(液相色谱三重四极杆/线性离子阱串联质谱)特有的 MRM 激发增强子离子扫描(EPI)相结合建立的多步模式,在离子阱内可富集二级子离子,即使较低浓度同样能够采集二级子离子碎片信息,实现定性的效果。经实验证实,在采集二级子离子的同时 MRM 数据可用于化合物的定量,定量结果偏差较小,准确度高。在本实验中前处理参照《动物源性食品中硝基呋喃类药物代谢物残留量检测方法高效液相色谱串联质谱法》(GB/T21311-2007)[7],处理后样液经质谱检测,其结果证实了利用 QTRAP 质谱能够实现一次进样得到硝基呋喃类药物同时定性定量结果,极大的简化了检验过程,并且在准确定性的基础上得到精确定量结果。
1 实验部分 1.1 仪器与试剂Shimadzu20AD 高效液相色谱仪(Shimadzu 公司); AB SCIEX4000QTRAP 三重四极杆/线性离子阱串联质谱仪,配有电喷雾离子源(TurboV,ABSCIEX); 高速组织捣碎机(德国 IKA 公司); 旋转浓缩仪(日本 EYELA 公司); Milli - Q 超纯水设备(美国 Millipore 公司); 漩涡混合器(德国 IKA 公司); 恒温水浴振荡器(江苏金坛精达仪器制造厂); 超声清洗仪(美国 BRANSON 公司); 低温高速离心机(湖南湘仪实验室仪器开发有限公司); 微孔滤膜为0.22μm。
AOZ、AMOZ、SEM 和 AHD 购自 Sigma 公司; 乙酸铵、2-硝基苯甲醛、乙酸乙酯、乙腈、甲醇等均为色谱纯; 其他试剂均为分析纯; 水为超纯水。
标准品储备液均用甲醇配制,4% 避光保存(有效期3个月); 工作液用水由储备液稀释,现用现配。
1.2 色谱条件色谱柱:Phenomenex Luna C18柱(150×2.0mm,3μm); 柱温:30℃ ; 进样体积:5μL; 流动相:A:含1mM 乙酸铵水溶液,B:含1mM 乙酸铵甲醇溶液;梯度洗脱。洗脱程序:0~6min,50% A 线性减少到25% ,保持2min; 至8.5min 线性增加到50% A,平衡5min。流速:200μL/min。
1.3 质谱条件多反应监测(MRM)扫描模式,ESI 离子源; CurtainGas 流量为30mL/min,Gas1流量为50mL/min,Gas2流量为50mL/min,辅助加热气温度为600℃,涉及气体均为氮气; 去簇电压(DP)和碰撞能量(CE)等质谱参数见表 1。利用信息依赖采集设定阈值5×107,选择动态背景扣除,在 Include 中添加呋喃唑酮、呋喃它酮代谢物 AOZ、AMOZ 母离子 m/z信息,此处阈值为1000。EPI 扫描范围:50~350Da; 扫描速率:20000Da/s; 离子阱填充时间:20ms,源参数与 MRM 模式一致,DP:70; CE:25;CXP:13。
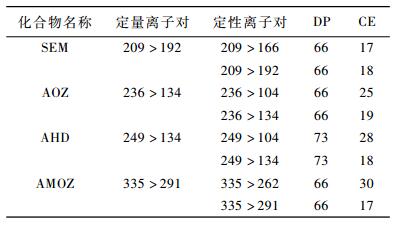
1.4 标准品二级碎片
设定碰撞能量 CE 为25,CES 为5,这样得到的谱图分别由能量20、25、30轰击母离子,3张谱图叠加后为标准品的二级碎片谱图,将其添加到标准谱库中。
1.5 样品处理称取约2g 试样(精确至0.01g)于50mL 塑料离心管中,加入10mL 甲醇—水混合溶液(1∶1,V/V),振荡10min 后,以4000r/min 离心5min,弃去液体。残留物中加入0.2mol/L 盐酸10mL ,用均质器以10000r/min 均质1min 后,加入0.1mol/L邻硝基苯甲醛溶液100μL,涡动混合30s 后,再振荡30min,置37℃恒温箱中过夜(16h)反应。
取出样品,冷却至室温,加入0.3mol/L 磷酸钾2mL(按照添加磷酸钾溶液体积的10倍加入盐酸溶液体积),用2.0mol/L 氢氧化钾调 pH7.4(±0.2后),再加入乙酸乙酯10~20mL(乙酸乙酯加入体积与盐酸溶液体积一致),振荡提取10min后,以10000r/min 离心10min,收集乙酸乙酯层。残留物用10~20mL 乙酸乙酯再提取1次,合并乙酸乙酯层。收集液在40℃下用 N2吹干,残渣用0.1% 甲酸水溶液1mL 溶解,再用乙腈饱和的正己烷3mL 分两次液液萃取,去除脂肪。下层水相过0.20μm 微孔滤膜过滤后,取10μL 供仪器测定。
2 检测方法的建立 2.1 质谱方法的建立与传统建立 MRM 模式类似,首先利用浓度为100ng/mL 的标准品优化化合物,确认化合物的准分子离子峰,子离子扫描(PI)找到丰度较高化合物的准分子离子碎片,通过优化去簇电压、碰撞能量得到碎片离子最优条件。连接色谱柱建立良好的液相条件,得到4种代谢物的色谱峰高、峰面积、离子比率、保留时间(图 1),用于化合物定量、初步定性确认。

|
| 图 1 硝基呋喃类药物代谢物的色谱图 |
2.2 样品测试
按照1.5中介绍步骤对样品前期处理,得到尽可能纯净的样液,放置进样盘固定位置,经液相色谱质谱测试。结果表明 AHD、AMOZ 存在干扰(图 2)。对比样品和标准溶液中 AHD、AMOZ 色谱峰,两者保留时间接近,分别相差0.1min、0.05min,而且由于存在基质干扰,可能导致保留时间略有偏差,所以无法根据保留时间区分化合物,依据以往离子比例均难以区分化合物。实验过程中,由于 AOZ、SEM 未受到基质干扰,所以研究过程中主要以 AHD、AMOZ 为对象,对比不同仪器采集模式得到的数据。

|
| 图 2 样品中硝基呋喃类药物代谢物及干扰的 TIC 色谱图 |
图 3中展示了 AHD 和 AMOZ 的相关 MRM 提取子离子对谱图和 EPI 扫描模式得到增强子离子全谱。其中 a1、a2、b1、b2分别对应保留时间为2.38min、2.43min、3.96min、4.06min MRM 离子对提取质谱图。对比 a1、b1两对 MRM 离子对比例,很难进行区分,同样 a2、b2无法确认化合物归属。运行子离子扫描模式(PI)分析采集数据没有得到有意义的结果。为准确判定性化合物的归属,需要借助离子阱类质谱仪,在化合物低浓度时借助离子阱的富集功能得到高质量有价值的质谱图。实验中选择 QTRAP 类质谱,除具有传统三重四极杆功能外,能够同时实现离子阱的富集作用,运行 MRM→IDA→EPI 复合扫描模式,实现一次进样得到化合物定性确认信息以及定量准确信息。当满足要求的离子进入 Q3时信息依赖性采集(IDA)实现增强子离子扫描(EPI),在线性离子阱内富集,得到成幂指数增长的子离子信号,可用于化合物二级子离子对比,以确认化合物归属。实验中分别用高、中、低3种碰撞能量对两种化合物进行 EPI 扫描,采集增强的子离子谱图通过3张谱图的叠加与标准品相同条件下叠加的二级子离子进行对比,以保证采集的数据更加准确、可靠。

|
| a1 AMOZ 标准溶液 MRM 提取离子流图; a2 AHD 标准溶液 MRM 提取离子流图; b1 AMOZ 样品中 MRM 提取离子流图; b2 AHD 样品中 MRM 提取离子流图; c1 AMOZ 标准溶液 EPI 增强子离子全谱; c2 AHD 标准溶液 EPI 增强子离子全谱; d1 AMOZ 样品中 EPI 增强子离子全谱; d2 AHD 样品中 EPI 增强子离子全谱。 图 3 HD 和 AMOZ 的相关谱图 |
图 3中 c1、d1分别代表保留时间为2.43min和2.38min 化合物经过富集的二级子离子谱图,图中可以清楚的对比出两者不同,与标准谱库进行对比,识别出保留时间在2.38min 的化合物为 AHD。同样 c2、d2分别代表保留时间为3.96min 和4.06min化合物经过富集的二级子离子谱图,经确认保留时间在4.06min 的化合物为 AMOZ。
以上结果说明运行 MRM→IDA→EPI 复合扫描模式针对低浓度化合物的识别与确认有着非常大的作用,并且操作简单,为除常规 MRM 两对离子对比例初步定性以外提供一条高效、识别率高、可信度高的定性模式,避免假阳性结果出现。
3 结果与讨论 3.1 复合扫描模式线性利用 MRM→IDA→EPI 复合扫描模式中化合物离子对信息对 AHD、AOZ、SEM、AMOZ4种化合物不同浓度空白基质加标样品采集得到的离子对色谱峰进行积分,线性范围0.01~5.0μg/kg,得到4种化合物的标准曲线(图 4)。

|
| 图 4 硝基呋喃及其代谢物标准曲线 |
3.2 结果重现性
实验中针对4种化合物运行利用 MRM→ IDA→EPI 复合扫描模式采集的色谱峰峰面积重现性实验,得到化合物的重现性均非常好,相对标准偏差均小于8% 。在0.01、0.1和0.5μg/kg3个添加水平下的平均回收率为76.3~97.6% ,相对标准偏差(CV%)为1.36~5.09; 化合物空白基质加标得出的检出限、检测限以及检测限时信噪比(S/N,按照噪音3倍标准偏差计算)、回收率(%)、相对标准偏差(重复采集6次)数据见表 2。表中可以看到4种化合物在检测限时的 S/N 均超过10,但是考虑到实际样品中可能存在干扰,适当放大检出限,此时同样满足硝基呋喃限量要求。
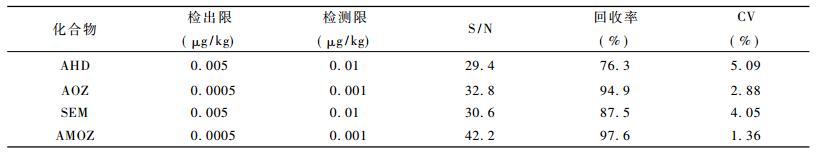
以浓度为0.01μg/kg SEM 原药的空白基质加标样品为例,连续6次进样,进样量为20μL,SEM的峰面积的重复性 CV% 为4.05% 。说明这种新型的复合扫描模式在能够有效确认识别化合物,对化合物同时进行定性、定量分析,准确度、重现性好,该方法适用于实验室日常监测工作。
4 结论研究建立水产品4种硝基呋喃及其代谢物的液相色谱串联质谱检测方法,采用 MRM→ IDA→EPI 复合扫描模式采集数据,能实现一次进样同时定性、定量分析,有效排除因基质干扰给实验结果带来的假阳性。方法在0.01~5.0μg/kg 浓度范围内,线性相关系数均>0.999,并且对化合物进行定性的同时不影响定量结果的准确度、重现性,满足国内外对硝基呋喃及其代谢的相关要求,为实验室检测该类化合物提供新的方法。
| [1] | 祝伟霞, 刘亚风, 梁炜. 动物性食品中硝基呋喃类药物残留检测研究进展[J]. 动物医学进展, 2010, 31(2): 99–102. |
| [2] | 曹鹏, 耿金培, 尹大路, 等. 高效液相色谱法同时测定饲料中的呋喃唑酮、呋喃西林、呋喃妥因、呋喃它酮药物残留量[J]. 山东农业大学学报(自然科学版), 2010, 41(3): 424–427. |
| [3] | 闫晓东, 王克宁, 张会彩, 等. 检测饲料中4种硝基呋喃类药物的简易高效液相色谱法[J]. 畜牧与兽医, 2010, 42(3): 86–89. |
| [4] | 王荣艳, 贾丽, 贾东芬, 等. UPLC-MS/MS法快速测定水产品中硝基呋喃类代谢物残留[J]. 分析试验室, 2010, 29(S1): 368–370. |
| [5] | 徐英江, 任传博, 田秀慧, 等. 海产品中硝基呋喃类原药的超高效液相色谱串联质谱测定[J]. 分析测试学报, 2010, 29(4): 327–330. |
| [6] | 张林田, 黄少玉, 陈建伟, 等. 高效液相色谱-串联质谱法测定水产品中硝基吠喃类代谢物[J]. 理化检验-化学分册, 2009, 45(9): 11–14. |
| [7] | 中国国家标准化管理委员会.GB/T21311-2007动物源性食品中硝基呋喃类药物代谢物残留量检测方法高效液相色谱串联质谱法[S].北京:中国标准出版社,2007. |