 2016, Vol. 36
2016, Vol. 36



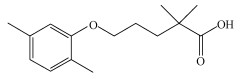
近年来, 由于在饮用水、污水处理厂以及地表水和地下水中被频繁检出, 药物和个人护理用品(PPCPs)作为一类新兴环境污染物日益受到人们的关注(Boyd et al., 2003; Vieno et al., 2007).该类物质难以生物降解, 在污水处理厂中不能被完全去除, 使得它们在自然水体中的含量不断累积(Ternes et al., 2004).吉非罗齐(Gemfibrozil, GEM)是一种临床常用的贝特类降血脂药物, 理化参数见表 1(Hassan et al., 2004; Westerhoff et al., 2005).其主要用来降低人体内甘油三酯和总胆固醇的水平, 减少冠心病的发病率.据报道, GEM在传统的污水处理工艺中的去除率较低, 大约集中在16%~46%之间(Stumpf et al., 1999).由于污水处理厂的不完全降解, GEM在各类水体中被频繁检测出来, 其中污水处理厂出水中检测浓度达到19.4 μg·L-1(Bendz et al., 2005), 在地表水中的浓度为0.75~1.5 μg·L-1(Sanderson et al., 2003), 在地下水中的浓度为0.751 μg·L-1(López Serna et al., 2013), 在饮用水中的浓度也达到0.07 μg·L-1(Fang et al., 2012).尽管没有研究表明GEM会对人体有直接毒性, 但多项研究表明其具有生态毒性, 会对大型溞和鱼类的生殖和生理产生不利影响(Skolness et al., 2012; Araujo et al., 2011).如何有效修复GEM带来的水体环境污染, 减弱其生态毒理效应, 成为众多学者关注的重点.
| 表 1 GEM的理化参数 Table 1 Physical and chemical properties of GEM |
传统的高级氧化工艺, 如UV/TiO2、UV/H2O2、UV/O3和Fenton法等均以产生强氧化性的羟基自由基(HO·)来氧化降解有机污染物(Oturan et al., 2014).近年来, 基于过硫酸盐活化的新型高级氧化工艺, 由于其自身的一些优势已经广泛应用于受污染土壤和地下水的原位化学氧化修复以及难降解有机废水的处理(Zhang et al., 2014).和HO·相比, 其所产生的硫酸根自由基(SO4·-)不仅在不同pH条件下都具有较高的氧化还原电位(E0=2.5~3.1 V), 而且主要通过电子转移途径来氧化降解有机物, 氧化反应更具有选择性(Guan et al., 2011; Neta et al., 1977).此外, SO4·-具有更长的半衰期, 可以增大与目标污染物的接触机会, 从而更高程度的矿化水体中的有机污染物(廖云燕等, 2014).目前, 活化过硫酸盐的方法主要有热活化、UV活化和过渡金属离子活化等(杨照荣等, 2013; Xie et al., 2015; 赵进英等, 2010).在各种活化方式中, 热活化过硫酸盐技术虽然在设备和操作上花费较高, 但由于其具有氧化能力强、反应条件温和、受pH影响小等优点, 已经广泛地应用于地下水以及工业废水中有机污染物的去除(杨照荣等, 2013; 邓靖等, 2014).此外, 由于系统操作简单, 该技术也应用于污染物降解机制的研究(Antoniou et al., 2010).
天然有机质(NOM)和碳酸氢盐(HCO3-)在自然水体中广泛存在, 并且已经被证实对基于SO4·-的高级氧化过程有显著影响(Ji et al., 2015).因此, 本研究以吉非罗齐为目标污染物, 研究其在热活化过硫酸盐体系中的降解机制.考察过硫酸盐初始浓度、温度、pH以及腐殖酸(HA)和HCO3-对GEM降解的影响, 并利用自由基清除实验鉴定活化体系中起主要作用的自由基, 探讨了不同pH条件下GEM的降解机制.最后利用HPLC-MS/MS技术鉴定活化体系中GEM主要的降解产物, 并推测其可能的降解路径.以期为热活化过硫酸盐技术在工程实际中的应用提供理论依据和技术支持.
2 材料与方法(Materials and methods) 2.1 试剂吉非罗齐(纯度>98%), TIC试剂公司;过硫酸钠、硫代硫酸钠、碳酸氢钠、硫酸和氢氧化钠均为分析纯, 国药集团化学试剂有限公司;腐植酸, 阿拉丁公司;乙腈、乙醇和叔丁醇均为色谱纯, 美国ACS恩科化学公司;实验过程用水均为超纯水.
2.2 实验方法用乙腈配置浓度为10 mmol·L-1的GEM储备液于棕色容量瓶中, 并放于4 ℃冰箱保存.每次实验准确移取1.0 mL GEM储备液于250 mL容量瓶中, 用高纯氮气吹干乙腈后用超纯水定溶(Smart2 Pure超纯水/纯水一体化系统, 德国TKA), 得到浓度为40 μmol·L-1的GEM反应液.取100 mL GEM反应液加入到250 mL锥形瓶中, 用1% NaOH和H2SO4调节溶液pH后置于恒温水浴装置, 待预热到指定温度, 加入Na2S2O8开始反应.每隔10 min, 取出1.0 mL样品并立即用1.0 mL的100 mmol·L-1Na2S2O3猝灭以终止反应, 然后用HPLC测定GEM的剩余浓度.每个时间点的样品至少设3个重复, 结果取平均值.
在该体系中分别考察不同过硫酸盐初始浓度、温度、pH、HA浓度和NaHCO3浓度条件下GEM的降解情况.自由基清除试验中, 用乙醇或叔丁醇作自由基清除剂, 在同样条件下考察GEM的降解情况.
2.3 分析测定方法GEM采用高效液相色谱仪(LC-20AT, SHIMADZU)进行测定, 色谱条件:色谱柱为C18色谱柱(Zorbax Eclipse XDB-C18, 2.1 mm×150 mm, 3.5 μm);流动相是甲醇-0.5%冰醋酸水溶液(75:25, V/V);检测器为光电二极管阵列检测器(SPD-M20A), 检测波长为276 nm, 流速为0.2 mL·.min-1, 进样量4 μL, 柱温为40 ℃.TOC采用TOC-VCPH(Shimadzu, 日本)进行测定.
GEM的降解产物由HPLC-MS/MS鉴定分析.40 μmol·L-1的GEM在温度为60 ℃、pH值为7.0、Na2S2O8浓度为1.5 mmol·L-1的溶液中反应90 min, 于旋转蒸发仪浓缩至1.5 mL, 转移至送检样品瓶中进行HPLC-MS/MS测定.测定仪器:高效液相色谱-离子肼质谱仪(Agilent 1100LC-MSD-Trap-XCT);色谱柱:Hypersil ODS色谱柱(250 mm×4.6 mm, 5 μm);进样量:10 μL;流动相组成:A水(0.2%冰醋酸), B甲醇.采用梯度洗脱:10%的B在15 min内上升到90%, 90%的B维持5 min;洗脱液流速:1.0 mL·min-1.MS部分采用电喷雾电离源(ESI), 检测模式为负离子检测;干燥气(N2)流速:10.0 L·min-1, 温度:350 ℃;碰撞气为氩气;离子源喷射电压:3500 V;雾化气(N2)压强为:40 psi;扫描范围:50~500 m·z-1.
3 结果(Results) 3.1 过硫酸盐初始浓度对GEM降解的影响由于过硫酸盐初始浓度能够直接影响SO4·-的平衡浓度, 所以在热活化体系中起着至关重要的作用(Ji et al., 2015).如图 1a所示, 过硫酸盐初始浓度显著影响GEM的降解.在没有加入过硫酸盐时, GEM很稳定, 几乎没有发生降解;随着过硫酸盐初始浓度的增加, GEM的降解速率显著加快.当过硫酸盐初始浓度为0.5 mmol·L-1时, 在60 min内GEM只有41.6%被去除;而当过硫酸盐初始浓度增加到3.0 mmol·L-1时, 同样的反应时间内GEM的去除率超过98%.在热活化过程当中, 过硫酸盐初始浓度越高, 反应体系中活化形成的氧化物种越多, 从而加速GEM的降解.
 |
| 图 1 过硫酸盐初始浓度对热活化过硫酸盐降解GEM的影响(T=60 ℃, pH=7.0, [GEM]0=40 μmol·L-1) Fig. 1 Effect of initial persulfate concentration on GEM degradation by heat-activated persulfate (T=60 ℃, pH=7.0, [GEM]0=40 μmol·L-1) |
GEM的降解过程可以用准一级反应动力学方程进行拟合:
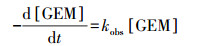
|
(1) |
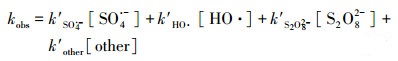
|
(2) |
式中, kobs为表观一级反应速率常数(min-1), [GEM]为GEM在某一时刻的浓度(μmol·L-1), k′为GEM与各种氧化物种的二级反应速率常数(L·mol-1·min-1).在热活化过硫酸盐体系中, SO4·-通常被认为是主要的氧化物种, 然而其他的氧化物种, 比如HO·等也可能在GEM的降解过程中发挥作用.图 1b为kobs随溶液中过硫酸盐初始浓度的变化情况, 从图中可知, 随着过硫酸盐初始浓度的增加, kobs随之加快, 并且呈现良好的线性关系.这说明GEM的降解速率和溶液中氧化剂的含量关系密切, 并且呈显著正相关.
3.2 温度对GEM降解的影响温度在热活化过硫酸盐体系降解污染物的过程当中也起着至关重要的作用.本研究考察40~70 ℃之间GEM的降解情况, 结果如图 2a所示.GEM的降解速率常数kobs随温度的升高而显著增加.40 ℃时, GEM的kobs为0.00156 min-1, 而当温度升高到70 ℃时, kobs增加到0.1356 min-1, 几乎增长了87倍.Ji等(2015)在用热活化过硫酸盐技术降解Atrazine时, 也发现温度从30 ℃升高到60 ℃时, kobs增长了114倍.随着反应溶液温度的升高, 过硫酸盐的分解速度加快, 反应液中自由基浓度增加, 从而使GEM的降解速率增加.此外, 根据热力学原理, 随着反应温度的升高, kobs也会随之升高.反应温度T和kobs两者之间的关系遵循阿仑尼乌斯方程:
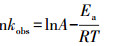
|
(3) |
 |
| 图 2 温度对热活化过硫酸盐降解GEM的影响(pH=7.0, [GEM]0=40 μmol·L-1, [persulfate]0=1.5 mmol·L-1) Fig. 2 Effect of temperature on GEM degradation by heat-activated persulfate (pH=7.0, [GEM]0=40 μmol·L-1, [persulfate]0=1.5 mmol·L-1) |
式中, Ea为反应的表观活化能(J·mol-1), R为气体常数(8.314 J·mol-1·K-1), A为指前因子(min-1), T为绝对温度(K-1).由图 2b可知, lnkobs随1/T的增加而线性减小, 两者之间的关系符合阿仑尼乌斯方程.由式(3)可以计算出反应的表观活化能为133.14 kJ·mol-1.
3.3 pH对GEM降解的影响溶液pH对热活化过硫酸盐降解GEM的影响如图 3所示.从图中可知, 在pH从5.0增加到10.0的过程当中, GEM的降解速率在逐渐降低.当pH从5.0增加到9.0时, 降解速率虽然在降低, 但GEM在60 min内仍然可以达到85%以上的去除率;而当pH从9.0升高到10.0时, GEM的去除率降到了69.0%.显然, 相对于碱性条件, 酸性和中性条件更有利于GEM的降解.
 |
| 图 3 pH对热活化过硫酸盐降解GEM的影响(T=60 ℃, [GEM]0=40 μmol·L-1, [persulfate]0=1.5 mmol·L-1) Fig. 3 Effect of pH on GEM degradation by heat-activated persulfate (T=60 ℃, [GEM]0=40 μmol·L-1, [persulfate]0=1.5 mmol·L-1) |
溶液pH在热活化体系当中起着复杂的作用.在酸性条件下, 过硫酸盐除了热活化产生SO4·-外(式(4)), 还会经历酸催化反应生成更多的SO4·-, 但高浓度的SO4·-会发生相互猝灭或者与过硫酸盐反应生成氧化能力更弱的自由基(式(5)~(7))(Anipsitakis et al., 2004; Clifton et al., 1989);在碱性条件下, SO4·-可以和H2O或者OH-反应生成HO·(式(8)~(9))(Zhao et al., 2013).此外, SO4·-与HO·和有机物的反应机制也不尽相同, 其中SO4·-主要通过电子转移的途径转化有机物;而HO·则优先加成到CC双键, 或者从C—H、N—H或O—H键上提取H, 可见SO4·-比HO·更具有选择性(Neta et al., 1977; Buxton et al., 1988).因此, pH可以通过改变活化体系生成的自由基类型来影响污染物的降解.
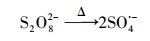
|
(4) |
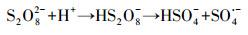
|
(5) |
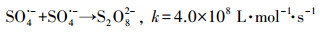
|
(6) |
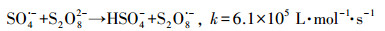
|
(7) |
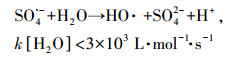
|
(8) |
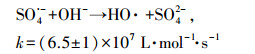
|
(9) |
碱性条件下GEM的降解效率低于酸性和中性条件, 正是由于不同pH条件下反应体系自由基的种类和活性不同造成的.在酸性和中性条件下, 体系主要的自由基是SO4·-, 在碱性条件下, 更多的SO4·-转化成了HO·.HO·虽然也具有较强的氧化能力, 但其氧化反应的无选择性减弱了与目标物种GEM的接触反应几率, 并最终降低GEM的降解效率.此外, GEM是离子型化合物, pKa为4.7, 在酸性条件下主要以分子形式存在, 在碱性条件下则更容易发生解离.随着溶液pH的升高, GEM的分子型比例下降, 离子型比例升高, GEM更多以离子形态存在, 更容易溶解于水中, 稳定性也增强, 不易被HO·等氧化物种降解, 从而使GEM在碱性条件下的去除效率降低.值得指出的是, 虽然酸性条件下会有更多SO4·-生成, 但pH=5.0时GEM的降解速率只是稍稍高于中性和弱碱性.这是因为体系中高浓度的SO4·-发生了相互猝灭或者转化成氧化能力较弱的自由基, 从而使GEM在酸性条件下并没有呈现较高的降解速率.
3.4 自然水体成分对GEM降解的影响本研究着重考察了HA和HCO3-对GEM降解的影响.HA由于分子结构中的富电子基团容易被SO4·-和HO·等亲电自由基攻击, 从而扮演者自由基清除剂的角色(Ghauch et al., 2013), 其对GEM降解的影响如图 4a所示.从图中可知, HA的加入明显抑制了GEM的降解, 而且抑制程度随HA浓度的增加而增强.未添加HA时, 60 min内GEM可以达到86%的去除率, 而当加入的HA增大到20 mg·L-1HA时, GEM的去除率降到了20%.这正是因为HA分子中的羟基、氨基等富电子活性基团和GEM竞争溶液中的SO4·-和HO·等氧化物种, 并最终抑制了GEM的降解.
 |
| 图 4 HA(a)和HCO3-(b)对热活化过硫酸盐降解GEM的影响(T=60 ℃, pH=7.0, [GEM]0=40 μmol·L-1, [persulfate]0=1.5 mmol·L-1) Fig. 4 Effect of HA (a) andHCO3-(b) on GEM degradation by heat-activated persulfate (T=60 ℃, pH=7.0, [GEM]0=40 μmol·L-1, [persulfate]0=1.5 mmol·L-1) |
HCO3-和CO32-是自然水体中常见的无机阴离子, 会对水体的碱度产生影响, 其中HCO3-的存在最为广泛.不同浓度HCO3-对GEM降解的影响如图 4b所示, 从图中可知, HCO3-对GEM的降解有强烈的抑制作用, 并且抑制程度随HCO3-浓度的增加而增强.当加入100 mmol·L-1HCO3-时, 60 min内GEM的去除率只有53.4%, 远远低于未加HCO3-时的86%.这一方面是因为HCO3-的加入, 增加了溶液的初始pH, 由上文可知, 碱性条件下GEM的降解效率要低于酸性和中性条件, 所以从改变溶液初始pH的角度, HCO3-的加入会抑制GEM的降解;另一方面是因为溶液一旦加入HCO3-, 系统便会产生HCO3-/CO32-(式10), 而HCO3-/CO32-作为SO4·-和HO·的猝灭剂, 可以转化生成HCO3·和CO3·-(式(11)~(15))(Liang et al., 2006; Bennedsen et al., 2012).HCO3·/CO3·-和苯酚、苯胺等富电子化合物有着较高的反应速率常数, 但它们和其他有机物的反应速率常数通常要比SO4·-和HO·低2~3个数量级(Acero et al., 2000).可见正是HCO3·和CO3·-对GEM较弱的氧化能力降低了GEM的降解速率.
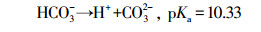
|
(10) |
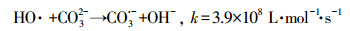
|
(11) |
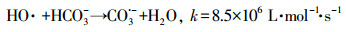
|
(12) |
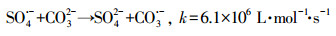
|
(13) |
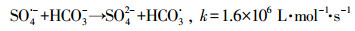
|
(14) |
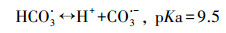
|
(15) |
为了探讨热活化过硫酸盐体系中GEM的降解机制, 反应体系在不同pH条件下的主要自由基被鉴定.本研究以乙醇(EtOH)和叔丁醇(TBA)作为自由基清除剂来鉴别不同pH条件下起主要作用的自由基.由式(16)~(19)可知, EtOH与SO4·-和HO·都具有较高的反应速率, 可以同时清除体系中的SO4·-和HO·;而TBA与SO4·-的反应速率要远远低于与HO·的反应速率, 故可以选择性清除HO·.利用两种自由基清除剂与SO4·-和HO·反应的不同活性, 可以区分反应液中的主要自由基类型(Ji et al., 2014; Zhou et al., 2013).
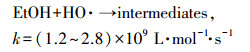
|
(16) |
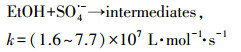
|
(17) |
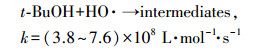
|
(18) |
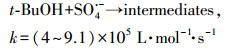
|
(19) |
由图 5可知, 在pH为5.0、7.0和10.0时, 加入60 mmol·L-1和600 mmol·L-1的EtOH均抑制了GEM的降解, 而且抑制作用随EtOH浓度的增加而增强, 这说明不同pH条件下GEM的降解都是活性自由基在起作用.而加入选择性清除HO·的TBA后, pH为5.0和7.0的条件下TBA对GEM降解的抑制作用都明显弱于EtOH的抑制作用, 这说明在酸性和中性条件下, 溶液中存在浓度较高的是SO4·-而不是HO·, SO4·-对GEM的降解起主导作用.当pH为10.0时, TBA的加入都强烈抑制了GEM的降解, 而且抑制程度几乎与加入EtOH时相当, 这说明在碱性条件下, HO·是体系主要的氧化物种, 其对GEM的降解起主导作用.
 |
| 图 5 EtOH和TBA作为自由基清除剂对热活化过硫酸盐降解GEM的影响(a) pH 5.0, (b) pH 7.0, (c) pH 10.0(T=60 ℃, [GEM]0=40 μmol·L-1, [persulfate]0=1.5 mmol·L-1) Fig. 5 Effect of EtOH and TBA as radical scavenger on GEM degradation by heat-activated persulfate: (a) pH 5.0, (b) pH 7.0, (c) pH 10.0 (T=60 ℃, [GEM]0=40 μmol·L-1, [persulfate]0=1.5 mmol·L-1) |
GEM在负离子检测模式下降解产物的总离子流色谱图(TIC)如图 6所示.从图中可知, 共检测到11种中间产物, 它们分别用P1~P11表示.根据总离子流色谱图和降解产物的二级质谱图信息, 可以推测降解产物的主要结构.GEM降解产物的质谱碎片信息以及推测的可能结构列于表 2.从表中可知, P1和P9是GEM苯环羟基加成后的产物;P5是苯环甲基被氧化为醛基的产物;P2则是苯环的羟基化和醛基化的产物;P6和P8是羟基化和醛基化的GEM侧链脱去羧基后的产物;P3则是苯环先经历羟基化和醛基化反应, 通过环化作用脱去两个氢后, 再脱去羧基后的产物;P4、P7、P10和P11都是苯环醚支链断裂的产物.
 |
| 图 6 GEM降解产物的总离子流图 Fig. 6 Total ion chromatogram (TIC) of GEM degradation products |
| 表 2 GEM降解产物的质谱碎片信息和推测的结构 Table 2 Mass spectrometry information and proposed structure of the degradation products of GEM |
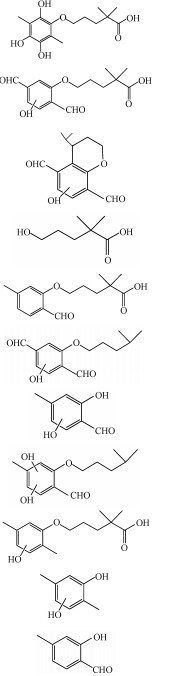
由此推测GEM在热活化过硫酸盐体系的降解路径主要有5条, 如图 7所示.路径一为苯环的羟基化反应, GEM苯环的大π电子体系和醚支链的供电子效应, 使得苯环的羟基加成反应容易发生(Song et al., 2008), 从而形成单羟基化的中间产物P9和多羟基化中间产物P1.路径二是苯环甲基的醛基化反应, 苯环甲基上HO·的加成和H提取形成碳中心自由基, 随后该自由基被O2氧化成甲醛, 最终形成醛基化的中间产物P5.此外, GEM苯环羟基化和醛基化反应也形成了中间产物P2.路径三是GEM侧链上的羧基在自由基进攻下的脱羧反应, 最终形成中间产物P3、P6和P8.路径四为苯环侧链的环化作用, 中间产物P2苯环支链经过环化作用, 脱去两个H, 并最终经过脱羧反应形成中间产物P3.路径五为ipso取代机制, HO·和SO4·-首先进攻和醚支链相连的芳香环碳原子(ipso位), 形成共振稳定的碳中心自由基, 该自由基进一步发生O-脱烷基反应, 使得醚氧和苯环之间键的断裂(Lam et al., 2005), 形成中间产物P4、P7、P10和P11.从以上分析中可以发现, GEM降解转化成中间产物后, 并没有发生进一步的降解.TOC测定实验也表明, GEM反应前后的矿化率只有16.5%, 这进一步证实大部分GEM都转化为中间产物, 而没有彻底矿化.
 |
| 图 7 GEM在热活化过硫酸盐体系中可能的降解路径 Fig. 7 Possible degradation pathways of GEM in heat-activated persulfate oxidation process |
由于我们研究的是中性条件下GEM在热活化过硫酸盐体系中的降解产物和可能的降解路径, 而从自由基清除实验可知, 中性条件下对降解起主导作用的是SO4·-, 所以以上降解产物应该主要是SO4·-参与降解的结果.HO·可以通过电子转移、脱氢和加成反应来降解有机物, 而SO4·-更倾向于电子转移的途径来转化降解有机物, 更具有选择性.从降解路径的分析中也可以发现, 苯环的羟基化可能是HO·参与降解的结果, 而以上5种降解路径都涉及到电子转移, 这进一步证实了中性条件下SO4·-降解的主导作用.此外, 五种降解路径中, 无论是羟基化还是醛基化的中间产物, 最终要完全降解, 都要经历醚支链的断裂, 断裂后的中间产物在自由基的作用下, 进一步发生降解, 才能最终矿化为CO2和H2O.所以醚支链的断裂可能是GEM最主要的降解路径.
Cermola等(2005)在研究模拟太阳光下GEM的光化学转化时, 也发现了苯环甲基的醛基化反应, 并通过核磁共振证实苯环邻位甲基更容易发生醛基化反应.Yurdakal等(2007)在研究GEM的TiO2光催化降解时发现醚支链的断裂是GEM的主要降解路径.Razavi等(2009)在研究自由基诱导下GEM的降解机制时, 发现了醚支链的断裂、苯环的羟基化反应以及苯环侧链的环化作用等降解路径, 本研究结果与其结论相一致.
4 结论(Conclusions)1) 热活化过硫酸盐技术可以有效降解水中的GEM, 其降解过程符合准一级反应动力学规律.
2) 增加过硫酸盐初始浓度和升高温度都可以显著提高GEM的降解速率常数.自然水体中的HA和HCO3-对GEM的降解有明显的抑制作用.相对于碱性条件, 酸性和中性条件更有利于GEM的降解.
3) 自由基清除实验表明, 在酸性和中性条件下, SO4·-对GEM的降解起主导作用, 而在碱性条件下, HO·成为体系主要的氧化物种.
4) HPLC-MS/MS分析结果表明, GEM主要的降解路径涉及苯环的羟基化和醛基化反应、苯环侧链的环化作用和脱羧反应以及醚支链的断裂.
| [${referVo.labelOrder}] | Acero J L, Stemmler K, Von Gunten U. 2000. Degradation kinetics of atrazine and its degradation products with ozone and OH radicals: a predictive tool for drinking water treatment[J]. Environmental Science & Technology , 34 (4) : 591–597. |
| [${referVo.labelOrder}] | Anipsitakis G P, Dionysiou D D. 2004. Radical generation by the interaction of transition metals with common oxidants[J]. Environmental Science & Technology , 38 (13) : 3705–3712. |
| [${referVo.labelOrder}] | Antoniou M G, Armah A, Dionysiou D D. 2010. Degradation of microcystin-LR using sulfate radicals generated through photolysis, thermolysis and e- transfer mechanisms[J]. Applied Catalysis B: Environmental , 96 (3) : 290–298. |
| [${referVo.labelOrder}] | Araujo L, Villa N, Camargo N, et al. 2011. Persistence of gemfibrozil, naproxen and mefenamic acid in natural waters[J]. Environmental Chemistry Letters , 9 (1) : 13–18. DOI:10.1007/s10311-009-0239-5 |
| [${referVo.labelOrder}] | Bendz D, Paxeus N A, Ginn T R, et al. 2005. Occurrence and fate of pharmaceutically active compounds in the environment, a case study: Höje River in Sweden[J]. Journal of Hazardous Materials , 122 (3) : 195–204. DOI:10.1016/j.jhazmat.2005.03.012 |
| [${referVo.labelOrder}] | Bennedsen L R, Muff J, Søgaard E G. 2012. Influence of chloride and carbonates on the reactivity of activated persulfate[J]. Chemosphere , 86 (11) : 1092–1097. DOI:10.1016/j.chemosphere.2011.12.011 |
| [${referVo.labelOrder}] | Boyd G R, Reemtsma H, Grimm D A, et al. 2003. Pharmaceuticals and personal care products (PPCPs) in surface and treated waters of Louisiana, USA and Ontario, Canada[J]. Science of the Total Environment , 311 (1) : 135–149. |
| [${referVo.labelOrder}] | Buxton G V, Greenstock C L, Helman W P, et al. 1988. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/·O-in aqueous solution[J]. Journal of Physical and Chemical Reference data , 17 (2) : 513–886. DOI:10.1063/1.555805 |
| [${referVo.labelOrder}] | Cermola M, DellaGreca M, Iesce M R, et al. 2005. Phototransformation of fibrate drugs in aqueous media[J]. Environmental Chemistry Letters , 3 (1) : 43–47. DOI:10.1007/s10311-005-0112-0 |
| [${referVo.labelOrder}] | Clifton C L, Huie R E. 1989. Rate constants for hydrogen abstraction reactions of the sulfate radical, SO4·- Alcohols[J]. International Journal of Chemical Kinetics , 21 (8) : 677–687. DOI:10.1002/(ISSN)1097-4601 |
| [${referVo.labelOrder}] | 邓靖, 冯善方, 马晓雁, 等.2014. 热活化过硫酸盐降解水中卡马西平[J]. 化工学报 , 2014, 66 (1) : 410–418. |
| [${referVo.labelOrder}] | Fang Y, Karnjanapiboonwong A, Chase D A, et al. 2012. Occurrence, fate, and persistence of gemfibrozil in water and soil[J]. Environmental Toxicology and Chemistry , 31 (3) : 550–555. DOI:10.1002/etc.v31.3 |
| [${referVo.labelOrder}] | Ghauch A, Ayoub G, Naim S. 2013. Degradation of sulfamethoxazole by persulfate assisted micrometric Fe0 in aqueous solution[J]. Chemical Engineering Journal , 228 : 1168–1181. DOI:10.1016/j.cej.2013.05.045 |
| [${referVo.labelOrder}] | Guan Y H, Ma J, Li X C, et al. 2011. Influence of pH on the formation of sulfate and hydroxyl radicals in the UV/peroxymonosulfate system[J]. Environmental Science & Technology , 45 (21) : 9308–9314. |
| [${referVo.labelOrder}] | Hassan H B, Kata M, Eräs I, et al. 2004. Preparation and investigation of inclusion complexes containing gemfibrozil and DIMEB[J]. Journal of Inclusion Phenomena and Macrocyclic Chemistry , 50 (3/4) : 219–225. |
| [${referVo.labelOrder}] | Ji Y, Dong C, Kong D, et al. 2015. Heat-activated persulfate oxidation of atrazine: Implications for remediation of groundwater contaminated by herbicides[J]. Chemical Engineering Journal , 263 : 45–54. DOI:10.1016/j.cej.2014.10.097 |
| [${referVo.labelOrder}] | Ji Y, Ferronato C, Salvador A, et al. 2014. Degradation of ciprofloxacin and sulfamethoxazole by ferrous-activated persulfate: implications for remediation of groundwater contaminated by antibiotics[J]. Science of the Total Environment , 472 : 800–808. DOI:10.1016/j.scitotenv.2013.11.008 |
| [${referVo.labelOrder}] | Lam M W, Young C J, Mabury S A. 2005. Aqueous photochemical reaction kinetics and transformations of fluoxetine[J]. Environmental Science & Technology , 39 (2) : 513–522. |
| [${referVo.labelOrder}] | Liang C, Wang Z S, Mohanty N. 2006. Influences of carbonate and chloride ions on persulfate oxidation of trichloroethylene at 20℃[J]. Science of the Total Environment , 370 (2) : 271–277. |
| [${referVo.labelOrder}] | 廖云燕, 刘国强, 赵力, 等.2014. 利用热活化过硫酸盐技术去除阿特拉津[J]. 环境科学学报 , 2014, 34 (4) : 931–937. |
| [${referVo.labelOrder}] | López Serna R, Jurado A, Vázquez-Suñé E, et al. 2013. Occurrence of 95 pharmaceuticals and transformation products in urban groundwaters underlying the metropolis of Barcelona, Spain[J]. Environmental Pollution , 174 : 305–315. DOI:10.1016/j.envpol.2012.11.022 |
| [${referVo.labelOrder}] | Neta P, Madhavan V, Zemel H, et al. 1977. Rate constants and mechanism of reaction of sulfate radical anion with aromatic compounds[J]. Journal of the American Chemical Society , 99 (1) : 163–164. DOI:10.1021/ja00443a030 |
| [${referVo.labelOrder}] | Oturan M A, Aaron J J. 2014. Advanced oxidation processes in water/wastewater treatment: principles and applications[J]. A review [J]. Critical Reviews in Environmental Science and Technology , 44 (23) : 2577–2641. DOI:10.1080/10643389.2013.829765 |
| [${referVo.labelOrder}] | Razavi B, Song W, Cooper W J, et al. 2009. Free-radicaL-induced oxidative and reductive degradation of fibrate pharmaceuticals: kinetic studies and degradation mechanisms[J]. The Journal of Physical Chemistry A , 113 (7) : 1287–1294. DOI:10.1021/jp808057c |
| [${referVo.labelOrder}] | Sanderson H, Johnson D J, Wilson C J, et al. 2003. Probabilistic hazard assessment of environmentally occurring pharmaceuticals toxicity to fish, daphnids and algae by ECOSAR screening[J]. Toxicology Letters , 144 (3) : 383–395. DOI:10.1016/S0378-4274(03)00257-1 |
| [${referVo.labelOrder}] | Skolness S Y, Durhan E J, Jensen K M, et al. 2012. Effects of gemfibrozil on lipid metabolism, steroidogenesis, and reproduction in the fathead minnow (Pimephales promelas)[J]. Environmental Toxicology and Chemistry , 31 (11) : 2615–2624. DOI:10.1002/etc.v31.11 |
| [${referVo.labelOrder}] | Song W, Cooper W J, Mezyk S P, et al. 2008. Free radical destruction of β-blockers in aqueous solution[J]. Environmental science & technology , 42 (4) : 1256–1261. |
| [${referVo.labelOrder}] | Stumpf M, Ternes T A, Wilken R D, et al. 1999. Polar drug residues in sewage and natural waters in the state of Rio de Janeiro, Brazil[J]. Science of the Total Environment , 225 (1) : 135–141. |
| [${referVo.labelOrder}] | Ternes T A, Joss A, Siegrist H. 2004. Peer reviewed: scrutinizing pharmaceuticals and personal care products in wastewater treatment[J]. Environmental Science & Technology , 38 (20) : 392A–399A. |
| [${referVo.labelOrder}] | Vieno N M, Härkki H, Tuhkanen T, et al. 2007. Occurrence of pharmaceuticals in river water and their elimination in a pilot-scale drinking water treatment plant[J]. Environmental Science & Technology , 41 (14) : 5077–5084. |
| [${referVo.labelOrder}] | Westerhoff P, Yoon Y, Snyder S, et al. 2005. Fate of endocrine-disruptor, pharmaceutical, and personal care product chemicals during simulated drinking water treatment processes[J]. Environmental Science & Technology , 39 (17) : 6649–6663. |
| [${referVo.labelOrder}] | Xie P, Ma J, Liu W, et al. 2015. Removal of 2-MIB and geosmin using UV/persulfate: Contributions of hydroxyl and sulfate radicals[J]. Water research , 69 : 223–233. DOI:10.1016/j.watres.2014.11.029 |
| [${referVo.labelOrder}] | 杨照荣, 崔长征, 李炳智, 等.2013. 热激活过硫酸盐降解卡马西平和奥卡西平复合污染的研究[J]. 环境科学学报 , 2013, 33 (1) : 98–104. |
| [${referVo.labelOrder}] | Yurdakal S, Loddo V, Augugliaro V, et al. 2007. Photodegradation of pharmaceutical drugs in aqueous TiO2 suspensions: mechanism and kinetics[J]. Catalysis Today , 129 (1) : 9–15. |
| [${referVo.labelOrder}] | Zhang B T, Zhang Y, Teng Y, et al. 2014. Sulfate Radical and its Application in Decontamination Technologies[J]. Critical Reviews in Environmental Science and Technology , 45 (16) : 1756–1800. |
| [${referVo.labelOrder}] | Zhao D, Liao X, Yan X, et al. 2013. Effect and mechanism of persulfate activated by different methods for PAHs removal in soil[J]. Journal of Hazardous Materials , 254 : 228–235. |
| [${referVo.labelOrder}] | 赵进英, 张耀斌, 全燮, 等.2010. 加热和亚铁离子活化过硫酸钠氧化降解4-CP的研究[J]. 环境科学 , 2010, 31 (5) : 1233–1238. |
| [${referVo.labelOrder}] | Zhou L, Zheng W, Ji Y, et al. 2013. Ferrous-activated persulfate oxidation of arsenic (III) and diuron in aquatic system[J]. Journal of Hazardous Materials , 263 : 422–430. DOI:10.1016/j.jhazmat.2013.09.056 |