 2016, Vol. 36
2016, Vol. 36



在典型碳酸盐岩发育地区, 岩溶地下河系统是极其重要的水资源储存空间, 满足了当地人群生活、生产和生态的基本需求, 如我国西南岩溶地区(袁道先等, 2007).然而, 当前岩溶地下河遭受硝酸盐污染的环境问题十分普遍.我国西南地区2836条岩溶地下河面临着变成“下水道”的实际威胁(袁道先等, 2007), 对地表水体的污染也造成了影响(薛禹群等, 2009).郭芳等(2002)的调查表明, 广西境内32条典型岩溶地下河硝酸盐浓度明显上升.人们通过同位素示踪研究(Liu et al., 2006; Jiang et al., 2009; Li et al., 2010)发现, 我国西南岩溶地下河硝酸盐污染与农业生产中过量施用化肥及农村废弃物不适当处置有密切关系.
反硝化作用已经被认为是修复地下水硝酸盐污染的有效路径, 在可利用碳源存在条件下具有很高的修复效果.例如, 王珍等(2008)利用锯木和零价铁作为碳源, 发现硝酸盐去除率达到100%;宋晓薇等(2013)用乙醇为碳源, 发现硝酸盐去除率达到99%;国外学者用液态碳源如甲醇、醋酸盐、葡萄糖、乙醇等, 发现硝酸盐去除率均在95%以上(Her et al., 1995; Gómez et al., 2000; Salminen et al., 2014; Carrey et al., 2014).然而, 大多数报道是在多孔介质条件下针对碳源选择而展开的.对于岩溶含水系统, 尤其是地下河系统, 有关利用反硝化作用去除硝酸盐的研究很少见.由于岩溶地下河具有特殊的管道结构和管道流特征, 与多孔介质相比, 其中的反硝化效果具有不确定性.针对岩溶地下河硝酸盐污染, 其修复策略也将是人们关注的问题.因此, 探索岩溶地下河中反硝化作用效果, 对岩溶水资源保护与利用具有积极意义.
由于真实岩溶地下河发育非均质性、环境许可制度及人为干扰因素复杂性, 野外开展污染物输入和输出试验十分困难.本次研究在桂林寨底地下河野外调查的基础上(陈余道等, 2013), 利用天然沉积的碳酸盐岩制作几何管道模型, 在实验室开展人工流条件下的硝酸盐生物去除实验研究, 旨在认识并评价岩溶地下河管道中反硝化作用效果, 分析其主要的影响因素.
2 材料与方法(Materials and methods) 2.1 主体实验 2.1.1 实验装置桂林是我国南方典型喀斯特发育地区, 境内的桂林寨底岩溶地下河系统是一个具有代表性的科研基地, 主要出露地层是泥盆纪和石炭纪的碳酸盐岩.最近, 在该地区响水岩-寨底泉地下河(直线距离约2.8 km), 利用荧光素钠作为示踪剂开展了一次枯水径流示踪试验, 估测了地下河管道结构参数和水力参数(陈余道等, 2013).为进一步研究地下河中反硝化作用效果, 本次研究结合现场调查制作了实验主体装置—碳酸盐岩管道几何模型(图 1).选择天然碳酸盐岩作为材料,一是考虑它的真实性,二是因为它具有缓冲酸碱度(pH值)的性能.
 |
| 图 1 参照桂林寨底地下河制作的碳酸盐岩管道模型(入口P1与蠕动泵相连供投注溶液, 出口P8与荧光监测仪相接测量出水中示踪剂浓度;P2~P7是管道上的注射器取样口) Fig. 1 Sketch of the carbonate-conduit model according to the understanding of Guilin Zhaidi Subterranean River, China |
根据X衍射荧光分析(ZSX Primus Ⅱ, 日本), 碳酸盐岩的成分主要为CaO (44.375%)、SiO2 (12.219%)、MgO (0.672%)、Al2O3 (1.535%)和Fe2O3 (0.357%).模型构件包括8个不同内径(1~5 cm)的碳酸盐岩管、1个球状碳酸盐岩空腔(内径10 cm)和3个不锈钢连接管.模型总长度367.5 cm, 管道内壁表面积5602 cm2, 体积8672 cm3.进水口P1连接进水蠕动泵, 出水口P8连接一个瑞士制造的GGUN-FL荧光检测仪(Goldscheider et al., 2008).在进水端和出水端之间设置了6个管道内部取样孔(P2~P7), 开口向上, 与进水口P1之间的距离分别为28、89、180、218、263、320 cm.
2.1.2 投注溶液实验开始前约半个月, 用投注溶液代替了管道中浸泡的地下水.投注溶液由新鲜的岩溶地下水做母液、加入化合物硝酸钠和荧光素钠配置而成, 物理化学性质如表 1所示.其中, 新鲜地下水作为土著微生物来源, 硝酸盐作为研究污染物(目标浓度约100 mg·L-1), 非反应示踪剂荧光素钠(浓度约50 μg·L-1)被用来对照其它投注物质的浓度变化.为反映地下河富氧特征, 投注溶液中溶解氧含量趋近于当地岩溶地下河常见值.参照真实地下河管道流平均滞留时间, 投注溶液流入、流出管道模型的速度设定为50 mL·h-1, 在管道中的平均滞留时间约173 h(即7.2 d).
| 表 1 实验采用的地下水物理化学特征 Table 1 Physical-chemical characteristics of groundwater used in the experiments |
反硝化作用依赖于可利用电子供体的存在, 否则会抑制反硝化作用的出现(Rivett et al., 2008).为了排除可利用碳源不足对地下河反硝化作用的影响,本次研究在实验开始一段时间后持续添加乙醇,作为唯一可利用碳源.根据已有研究(Chen et al., 2008; 2013; 宋晓薇等, 2013), 乙醇是微生物嗜好的碳源, 是一种能够与水混溶的液体, 容易被定量, 这里不试图说明现场使用乙醇的前景.添加的乙醇随投注溶液一起进入管道, 目标浓度为300 mg·L-1.
2.2 辅助实验本次研究还利用了一个以前的砂柱实验结果, 来与主体实验对比反硝化作用的效果.该装置是一个上流式的有机玻璃圆柱反应器, 内径5 cm, 柱高120 cm, 全部被含水层细砂充填.X衍射荧光分析表明, 细砂的主要成分为:SiO2(77.73%)、Al2O3(11.046%)、Fe2O3(3.769%)、CaO(1.723%)和MgO(0.051%), 反映了浅层孔隙含水层的沉积物组成.与碳酸盐岩管道材质一样, 沉积物不含明显的可利用电子供体, 如还原态铁和硫.进水由同源的新鲜地下水经人工配制构成, 其中, 硝酸盐浓度200 mg·L-1, 乙醇浓度100 mg·L-1, 进水流量为45 mL·h-1, 柱内平均滞留时间约25 h.值得注意的是, 与主体实验比较, 辅助实验投注乙酸浓度是前者的2倍, 投注碳源浓度是前者的1/3, 装置内的平均滞留时间大约是前者的1/7.
2.3 采样与分析每天定时使用注射器采集碳酸盐管道进口和出口水样, 采样体积分别为21 mL, 按需要在实验期间沿P2~P7采集管道水样.对于辅助实验, 每小时采集砂柱进水和出水样品, 采样体积分别为10 mL.
使用气相色谱仪(Agilent6890N)分析乙醇, 用离子色谱仪(DIONEX ICS-1000 IC)分析硝酸盐和亚硝酸盐, 用德国产的WTW仪器(Multi 3420)分析溶解氧、pH、温度, 用德国默克化工生产的Calcium-test和Alkalinity-test检测测试药剂和测试重碳酸根浓度, 由GGUN-FL荧光检测仪自动监测荧光素钠示踪剂浓度.各物质的检测下限分别为:乙醇0.1 mg·L-1, 硝酸盐0.1 mg·L-1, 亚硝酸盐0.01 mg·L-1, 重碳酸根1 mg·L-1, 荧光素钠0.001 mg·L-1.气相色谱仪和离子色谱仪的检测方法见文献(陈余道等, 2014).
主体实验期间, 在出水口P8采集水样1 L, 用来检测微生物丰度.用荧光定量PCR仪(CFX Manager2.1Bio-rad, Hercules, 美国)测定单位水体积中16S rRNA和nirK基因拷贝数, 分别作为总细菌(Yaoa et al., 2006)和反硝化细菌(Henry et al., 2004)的指示丰度.
3 结果与讨论(Results and discussion)主体实验持续2184 h(91 d), 合计向管道中投注溶液体积为101.62 L, 与出水体积100.45 L和管道沿途取样体积1.17 L之和持平.投注溶液中溶解氧、硝酸盐和乙醇浓度变化如图 2所示.由图可知, 溶解氧浓度变化较平稳, 平均达8.3 mg·L-1;硝酸盐和乙醇浓度具有人为的波动, 平均值分别达99.1和285.9 mg·L-1.其中, 乙醇投注时间为第156~1488 h.
 |
| 图 2 进水中DO、NO3-和乙醇的浓度变化 Fig. 2 Concentrations of DO, nitrate and ethanol in the influent |
投注溶液沿管道迁移至出口, 其组分在出水中表现出了不同程度的浓度-质量变化.首先, 溶解氧和硝酸盐浓度在添加碳源后均发生了显著变化(图 3), 它们的浓度检出范围分别为8.1~1.2 mg·L-1和102.6~40.3 mg·L-1.相比之下, 作为非反应示踪剂, 荧光素钠浓度变化很小, 范围为45.5~51.6 μg·L-1.在添加碳源前(0~155 h), 溶解氧和硝酸盐的出水浓度与进水浓度相近, 变化小;随着碳源的持续添加, 出水中乙醇浓度逐渐增加并相对稳定, 而溶解氧和硝酸盐浓度则相继急剧下降.其中, 硝酸盐浓度的下降速率达到0.114 mg·L-1·h-1 (图 3 Fit 1), 硝酸盐在第732~1548 h期间平均浓度为49.2 mg·L-1;溶解氧在第456~1488 h期间的平均浓度为1.6 mg·L-1.
 |
| 图 3 出水中投注组分的浓度变化(Y表示硝酸盐浓度(mg·L-1), X表示时间(h)) Fig. 3 Concentration changes of the injected solutes in the effluent |
其次, 溶解氧和硝酸盐的质量也出现了显著变化(表 2).添加碳源前(0~155 h), 进水和出水中硝酸盐和溶解氧的质量变化很小, 相对误差分别为1.53%和0.83%;持续添加碳源期间(156~1488 h), 出水中硝酸盐质量比进水质量减少39.4%, 溶解氧质量减少70.2%, 而乙醇质量也减少34.1%.
| 表 2 溶解氧、硝酸盐和乙醇在进、出水中的质量变化统计 Table 2 Mass estimations of DO, nitrate and ethanol in the influent and effluent |
对照示踪剂的浓度变化, 出水中溶解氧和硝酸盐出现了浓度下降与质量衰减过程, 这与碳源的添加有密切联系.由此可以认为, 这些物质之间发生了化学反应, 即存在乙醇被氧化和被反硝化的过程.乙醇作为碳源, 也是电子供体, 氧气和硝酸盐作为电子受体, 微生物获得碳源和能源.反应过程可以用下列关系概括:
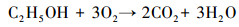
|
(1) |
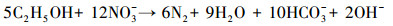
|
(2) |
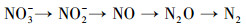
|
(3) |
实验中对反应产物包括亚硝酸盐和重碳酸根的监测也能证明上述反应的存在.如图 4所示, 在添加碳源后, 反应生成的二氧化碳导致出水重碳酸盐浓度快速增加, 最高达186 mg·L-1;亚硝酸盐成为反硝化作用的中间产物, 最高浓度达到5.24 mg·L-1.
 |
| 图 4 亚硝酸盐和重碳酸根的浓度变化 Fig. 4 Concentration changes of nitrite and bicarbonate ion |
除了地球化学信息外, 实验中期(约第960 h)管道水样的微生物检测得出, 总细菌丰度(16S rRNA基因丰度)为2.96×107 copies·L-1, 反硝化菌丰度(nirK基因丰度)为6.23×106 copies·L-1, 其比值(nirK/16S rRNA)为21.05%, 表明管道水中存在大量的微生物, 包括反硝化菌.但反硝化菌占总细菌数的比例不足25%.
3.2 反硝化作用的有限性有趣的是, 主体实验出现了一个以前多孔介质实验中没有发生的现象, 也就是在可利用碳源充足条件下硝酸盐没有得到完全去除.从图 3可以看出, 硝酸盐浓度在第1548 h后没有持续下降, 而是开始了上下波动.通过线性拟合这组波动数据, 可以得出一个近水平的线性方程(图 3 Fit 2), 斜率为0.001, 截距为49.2.质量统计(表 2)也表明, 在持续添加碳源期间(156~1488 h)硝酸盐质量减少率仅为39.4%.
在以前的辅助实验中, 尽管硝酸盐投注的目标浓度(200 mg·L-1)比主体实验(100 mg·L-1)高, 乙醇浓度(100 mg·L-1)比主体实验(300 mg·L-1)低, 但硝酸盐去除率、去除速率都大于主体实验.多孔介质中硝酸盐浓度呈未检出水平, 去除率大于99%, 反硝化动力学速率达10.8 mg·L-1·h-1(图 5).对比这些数据表明, 即使在碳源充足条件下, 碳酸盐岩管道中反硝化作用也受到了明显的约束.
 |
| 图 5 辅助实验中的硝酸盐和碳源乙醇的浓度变化(Y表示硝酸盐浓度(mg·L-1), X表示时间(h)) Fig. 5 Concentrations of nitrate and ethanol in the assistant experiment |
由于地下环境普遍存在反硝化菌, 通常能够影响反硝化作用效果的因素除了可利用电子供体(碳源)和电子受体浓度外, 还有环境条件, 如温度、pH值、营养物和介质空隙大小等(Rivett et al., 2008).对于硝酸盐污染的情形, 硝酸盐本身就是电子受体, 浓度高.下面将从碳源、溶解氧和环境因素方面进行讨论.
3.3.1 碳源(电子供体)一个氧化还原反应缺乏可利用电子供体, 反应是不能进行的, 这也是限制反硝化作用的固有因素.从本次研究可以看出, 没有碳源, 反硝化作用去除硝酸盐的效果不明显, 一旦添加了碳源, 硝酸盐被去除的效果明显增加.
3.3.2 溶解氧的影响作为自然环境中的电子受体, O2具有比其它电子受体(如NO3-)更强的争夺电子供体的能力.O2转化为H2O的氧化还原电位Eh值为+0.805 V, 而硝酸盐转化为N2和NO2-的Eh分别为+0.712 V和+0.404 V(Wiedemeier et al., 1999).在电子供体的利用上, 需氧呼吸优先, 其次为反硝化呼吸.
由于岩溶地下河系统的开放性及与地表水密切的水力联系, 水体中的溶解氧含量通常很高, 比如, 桂林寨底地下河出口处的溶解氧为8.0~8.3 mg·L-1.本次研究表明, 溶解氧衰减的启动时间比硝酸盐早, 明显下降的时间分别为第192~212 h和第384~396 h(图 3), 图 6也表明在水流路径上溶解氧浓度降低比硝酸盐提前, 首先在上游P2取样点出现了溶解氧和乙醇浓度的降低, 然后在下游P4取样点才出现硝酸盐浓度的降低.
 |
| 图 6 溶解氧、硝酸盐和乙醇浓度沿水流路径的变化 Fig. 6 Concentration changes of DO, nitrate and ethanol along the flow path |
溶解氧对反硝化作用的影响体现在利用优先碳源上.可以设想, 如果本次实验添加的碳源有限, 就可能出现碳源被需氧呼吸消耗而反硝化不能启动的情形.然而, 当碳源相对富余时, 溶解氧被利用使得浓度下降到一定程度, 反硝化作用将启动.在主体实验中, 硝酸盐浓度明显降低时, 溶解氧浓度大约为3.0 mg·L-1.
3.3.3 环境因素温度、pH和营养物的影响:主体实验进、出水温度为16.5~25.0 ℃, 进水pH值平均为8.08, 出水pH值平均值为7.89.这些值与我国南方岩溶地下河水体特征值基本一致, 不会影响微生物的代谢生长.对于营养物, 氮素本身也是营养成分.因此, 相对于溶解氧和碳源缺失这两个因素, 真实岩溶地下河中环境因素对反硝化的影响可以忽略.
空隙固体表面积和水体积比值的影响:岩溶地下河常被看成是管道流, 在储水空间大小方面与多孔介质有显著差别.管道储水空间大, 单位体积内固体表面积比多孔介质颗粒表面积小, 或者说, 固体表面积和水体积比值较小.根据已有文献, Harvey等(1984)认为不管是在污染含水层还是在未污染含水层, 大部分细菌是附着在固体颗粒表面的;Lehman等(2001)也认为附着在颗粒表面的生物量达到99%.由此推测, 岩溶地下河管道内生物量密度要比多孔介质环境低, 从而影响了管道内的反硝化作用, 使得管道内硝酸盐去除率较低.
4 结论(Conclusions)1)碳酸盐岩管道中存在反硝化作用的潜能, 通过反硝化菌的代谢可以去除水体中的硝酸盐.碳源是反硝化作用首要的影响因素, 没有可利用电子受体意味着反硝化作用难以出现.
2)碳酸盐岩管道流与多孔介质流不同:即使碳源充足的情况下, 反硝化作用程度也会受到限制, 硝酸盐去除仍然是有限的.其原因主要与管道空隙固体表面积与水体积比值小、管道流存在丰富的溶解氧有关, 而地下河系统中其它环境因素(如pH值和温度)影响不明显.
| [${referVo.labelOrder}] | Carrey R, Otero N, Vidal-Gavilan G, et al. 2014. Induced nitrate attenuation by glucose in groundwater: Flow-through experiment[J]. Chemical Geology , 370 : 19–28. DOI:10.1016/j.chemgeo.2014.01.016 |
| [${referVo.labelOrder}] | 陈余道, 程亚平, 王恒, 等.2013. 岩溶地下河管道流和管道结构及参数的定量示踪——以桂林寨底地下河为例[J]. 水文地质工程地质 , 2013, 40 (5) : 8–12. |
| [${referVo.labelOrder}] | 陈余道, 宋晓薇, 蒋亚萍, 等.2014. 岩溶地下河系统石灰石对BTEX的吸附动力学和热力学[J]. 地学前缘 , 2014, 21 (4) : 180–185. |
| [${referVo.labelOrder}] | Chen Y, Jiang Y, Zhu Y, et al. 2013. Fate and transport of ethanol blended dissolved BTEX hydrocarbons: A quantitative tracing study of a sand tank experiment[J]. Environmental Earth Science , 70 (1) : 49–56. DOI:10.1007/s12665-012-2102-4 |
| [${referVo.labelOrder}] | Chen Y D, Barker J F, Gui L. 2008. A strategy for aromatic hydrocarbon bioremediation under anaerobic conditions and the impacts of ethanol: A microcosm study[J]. Journal of Contaminant Hydrology , 96 (1) : 17–31. |
| [${referVo.labelOrder}] | Goldscheider N, Meiman J, Pronk M, et al. 2008. Tracer tests in karst hydrogeology and speleology[J]. International Journal of Speleology , 37 (1) : 27–40. DOI:10.5038/1827-806X |
| [${referVo.labelOrder}] | 郭芳, 姜光辉, 裴建国, 等.2002. 广西主要地下河水质评价及其变化趋势[J]. 中国岩溶 , 2002, 21 (3) : 195–201. |
| [${referVo.labelOrder}] | Gómez M A, González-López J, Hontoria-García E. 2000. Influence of electron donor on nitrate removal of contaminated groundwater in a denitrifying submerged filter[J]. Journal of Hazardous Materials , 80 (1) : 69–80. |
| [${referVo.labelOrder}] | Harvey R W, Smith R L, George L. 1984. Effect of organic contamination upon microbial distributions and heterotrophic uptake in a Cape Cod, Mass[J]. , aquifer [J]. Applied and Environmental Microbiology , 48 (6) : 1197–1202. |
| [${referVo.labelOrder}] | Henry S, Baudoin E, López-Gutiérrez J C, et al. 2004. Quantification of denitrifying bacteria in soils by nirK gene targeted real-time PCR[J]. Journal of Microbiological Methods , 59 (3) : 327–335. DOI:10.1016/j.mimet.2004.07.002 |
| [${referVo.labelOrder}] | Her J J, Huang J S. 1995. Influences of carbon source and C/N ratio on nitrate/nitrite denitrification and carbon breakthrough[J]. Bioresource Technology , 54 (1) : 45–51. DOI:10.1016/0960-8524(95)00113-1 |
| [${referVo.labelOrder}] | Jiang Y, Wu Y, Yuan D. 2009. Human impacts on Karst groundwater contamination deduced by coupled nitrogen with strontium isotopes in the Nandong subterranean river system in Yunan, China[J]. Environmental Science Technology , 43 (20) : 7676–7683. DOI:10.1021/es901502t |
| [${referVo.labelOrder}] | Lehman R M, Colwell F S, Bala G A. 2001. Attached and unattached microbial communities in a simulated basalt aquifer under fracture-and porous-flow conditions[J]. Applied and Environmental Microbiology , 67 (6) : 2799–2809. DOI:10.1128/AEM.67.6.2799-2809.2001 |
| [${referVo.labelOrder}] | Li S L, Liu C Q, Lang Y C, et al. 2010. Tracing the sources of nitrate in karstic groundwater in Zunyi, Southwest China: a combined nitrogen isotope and water chemistry approach[J]. Environmental Earth Science , 60 (7) : 1415–1423. DOI:10.1007/s12665-009-0277-0 |
| [${referVo.labelOrder}] | Liu C Q, Li S L, Lang Y C, et al. 2006. Using δ15N-and δ18O-values to identify nitrate sources in Karst ground water, Guiyang, Southwest China[J]. Environmental Science Technology , 40 (22) : 6928–6933. DOI:10.1021/es0610129 |
| [${referVo.labelOrder}] | Rivett M O, Buss S R, Morgan P, et al. 2008. Nitrate attenuation in groundwater: A review of biogeochemical controlling processes[J]. Water Research , 42 (16) : 4215–4232. DOI:10.1016/j.watres.2008.07.020 |
| [${referVo.labelOrder}] | 宋晓薇, 蒋亚萍, 陈余道, 等.2013. 2种生物反硝化法去除地下水中硝酸盐的研究[J]. 环境科学与技术 , 2013, 36 (5) : 108–111. |
| [${referVo.labelOrder}] | Salminen J M, Petäjäjärvi S J, Tuominen S M, et al. 2014. Ethanol-based in situ bioremediation of acidified, nitrate-contaminated groundwater[J]. Water Research , 63 : 306–315. DOI:10.1016/j.watres.2014.06.013 |
| [${referVo.labelOrder}] | Wiedemeier T H, Wilson J H, Kampbell D H. 1999. Technical protocol for implementing intrinsic remediation with long-term monitoring for natural attenuation of fuel contamination dissolved in groundwater, vol 1[R].San Antonio:TXAir Force Center for Environmental Excellence, Technology Transfer Division Brooks AFB |
| [${referVo.labelOrder}] | 王珍, 张增强, 唐次来, 等.2008. 生物-化学联合法去除地下水中硝酸盐[J]. 环境科学学报 , 2008, 28 (9) : 1839–1847. |
| [${referVo.labelOrder}] | 薛禹群, 张幼宽.2009. 地下水污染防治在我国水体污染控制与治理中的双重意义[J]. 环境科学学报 , 2009, 29 (3) : 474–481. |
| [${referVo.labelOrder}] | Yaoa S, Merwina I A, Abawib G S, et al. 2006. Soil fumigation and compost amendment alter soil microbial community composition but do not improve tree growth or yield in an apple replant site[J]. Soil Biology and Biochemistry , 38 (3) : 587–599. DOI:10.1016/j.soilbio.2005.06.026 |
| [${referVo.labelOrder}] | 袁道先, 薛禹群, 傅家谟, 等, 2007.防止我国西南岩溶地区地下河变成"下水道"的对策与建议[Z]., 中国科学院院士建议, (4): 1-14 |