 2016, Vol. 36
2016, Vol. 36



2. 中国科学院生态环境研究中心, 饮用水科学与技术重点实验室, 北京 100085
2. State Key Laboratory of Environment Aquatic Chemistry, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085
甲基砷是一类常见的有机砷,作为除草剂和杀菌剂被广泛使用,在天然水体中常被检出(Kitchin et al.,1984).典型的甲基砷包括一甲基砷(MMA,monomethylarsinic)和二甲基砷(DMA,dimethylarsinic),这些甲基砷进入人体后易损伤人体DNA而严重威胁人类健康(Dopp et al.,1984;Braman et al.,1973);在一定条件下,MMA和DMA有可能被还原成MMA(Ⅲ)和DMA(Ⅲ),它们的毒性会进一步增强(Petrick et al.,2000).因此对水体中甲基砷特别是饮用水中的甲基砷进行有效控制或去除显得非常必要.
近年来,甲基砷的去除方法集中在吸附法和光催化法.Shimizu等研究了MMA和DMA在铁/铝氧化物表面的吸附行为,结果表明在5 min内,MMA的去除率可达78%,而DMA的去除率仅15%(Shimizu et al.,2010);景传勇等考察了纳米晶-锐钛矿型TiO2 对各形态砷吸附效果和吸附机制,结果表明TiO2 对砷的吸附量大小顺序是 As(V)>MMA>DMA(Jing et al.,2005);徐忠厚等(2008)采用纳米 TiO2 光催化氧化降解DMA的机制研究.结果表明,在紫外光催化作用72 h后,仅有60%的DMA 转化成MMA及As(V)(Xu et al.,2008);Tsunenori Nakajima利用Al2O3负载的TiO2催化剂降解MMA和DMA,催化剂投量为1.0 g·L-1,反应时间长达10 h时,MMA和DMA才可被完全去除(Nakajima et al.,2005).Hu等对DMA的去除效果作了进一步研究,研究结果发现,常规的氧化剂KMnO4和NaClO对DMA的去除效率约30%,壳聚糖和粉末活性对DMA的去除率也不超过50%(Hu et al.,2015).DMA甲基化程度比MMA高,结构更为稳定,DMA更难被有效去除.
电芬顿是一种降解有机污染物的有效方法,电芬顿系统中阴极原位产生的H2O2可同步与金属离子或金属氧化物发生芬顿或类芬顿反应,产生的羟基自由基可快速降解甚至矿化目标污染物(肖华等,2004;Outran et al.,2005).例如,Guivarch等利用电芬顿法对有机磷杀虫剂进行降解,该杀虫剂的矿化率可达80%以上(Guivarch et al.,2003);Wang等采用电芬顿法矿化偶氮染料酸性红14,反应360 min,酸性红14的TOC去除率可达70%(Wang et al.,2005).但是利用电芬顿法去除水中甲基砷,特别是DMA鲜有报道,因此本研究中将制备新型负载Fe3C纳米粒子的炭纤维电芬顿催化剂,研究该催化剂电芬顿降解DMA的效果、影响因素及降解机制.
2 材料与方法(Materials and methods) 2.1 催化剂制备聚丙烯腈、N,N-二甲基甲酰胺(DMF)、乙酰丙酮铁、无水硫酸钠(Na2SO4)、硫酸(H2SO4)、氢氧化钠(NaOH)购自国药化学试剂公司,二甲基砷酸钠C2H6AsNaO2·3H2O(J &K,China)一甲基砷酸钠CH4AsNaO3·1.5H2O(Sigma,USA)、砷酸钠Na2HAsO4·7H2O(Sigma,USA)均为分析纯,活性炭纤维(ACF)由山东雪圣科技有限公司提供.实验所用水由 Milli-Q超纯水机制得(18.2 MΩ).
将1000 mg聚丙烯腈、1000 mg乙酰丙酮铁、10 mL N,N-二甲基甲酰胺置于50 mL烧杯中,利用磁力搅拌器混合搅拌均匀制得纺丝液,通过静电纺丝装置制备出负载铁的纳米纤维状结构,将聚合物纤维模板置于管式炉中在空气气氛下280 ℃预氧化2 h,并进一步在氮气气氛保护下850 ℃碳化1 h,最终得到负载纳米Fe3C粒子的炭纤维催化剂(Fe3C/CF).
2.2 电催化实验测试在Fe3C/CF电催化降解DMA的实验中,取 120 mL 浓度 5 mg·L-1的DMA储备液置于容积为 120 mL特制石英反应器中.阳极采用尺寸为4 cm × 5 cm RuO2/Ti 网状电极,阴极为相同尺寸活性炭纤维电极.电解质为0.05 mol·L-1的 Na2SO4,用 0.2 mol·L-1 H2SO4或0.2 mol·L-1NaOH调节溶液pH值,O2流量控制在40~60 mL·min-1.通电并投加一定量Fe3C/CF催化剂开始实验,分别在0、30、60、90、120、180、240、360 min取样,稀释后用0.22 μm膜过滤.电源为DH1718E-4 型直流双路跟踪稳压稳流电源(北京大华电子).
2.3 样品分析与材料表征采用高效液相色谱电感耦合等离子体质谱联用技术(HPLC-ICP-MS)测定DMA、MMA和As(V)浓度,检测条件为:Hamilton PRP-X100色谱柱(250 mm×4.1 mm,10 μm),柱温30 ℃,进样量20 μL,流动相为10 mmol·L-1(NH4)2HPO4缓冲溶液(用冰醋酸调节pH = 6),流速为等速,1.0 mL·min-1,RF入射功率1380 W,载气为高纯氩气,载气流速1.12 L·min-1,泵速0.3 r·s-1,检测质量数m/z 75(As)(Liu et al.,2013).采用电子自旋共振波谱仪(ESR)检测·OH的生成,使用的自由基加和试剂为DMPO.检测条件为:中心场强3511.940 C、扫描宽度为100.000 G、解析点1024、微波频率为9.857 GHz、功率2.301 mW(奚倩等,2014).
采用X射线粉末衍射仪(X′Pert PRO MPD,荷兰帕萨科分析仪器有限公司)对产物物相进行表征,通过扫描电子显微镜(SU-8020,日本日立公司)对产物形貌进行表征,利用比表面积测定仪(ASAP2000,Micromeritics 公司)测定产物的氮气吸附等温线、比表面积、孔径分布.
3 结果与讨论(Results and discussion) 3.1 催化剂表征图 1(b)为本研究中制备的催化剂的XRD谱图,产物的衍射峰与Fe3C标准卡片(图 1a)完全一致,表明成功制备了含Fe3C的催化材料.
 |
| 图 1 Fe3C/CF 催化剂的XRD谱图 Fig. 1 XRD pattern of Fe3C/CF |
图 2为碳化前后Fe3C/CF催化剂的SEM形貌图片及EDX元素分析结果.由图 2a可以看出,Fe3C/CF催化剂纺丝前驱体形貌呈光滑纤维状,纤维的长度达几十μm,而直径均匀分布在500~600 nm间.图 2b中经碳化处理制备的催化剂呈现出断裂的纤维状结构,长度分布从几μm到十几μm不等.从图 2c可以看出,碳纤维表面被大量纳米尺寸的颗粒物均匀包裹.EDX的检测结果发现这些纳米颗粒物的组成主要为碳和铁,铁含量约为8.43%.
 |
| 图 2 碳化前后 Fe3C/CF 催化剂的SEM 照片及EDX分析(a. Fe3C/CF 前驱体;b. Fe3C/CF的低倍数图片;c. Fe3C/CF的高倍数图片; d. Fe3C/CF的EDX结果) Fig. 2 SEM images of Fe3C/CF before and after carbonization of(a. Precursor of Fe3C/CF;b. Low-magnification image of Fe3C/CF; c.High-magnification image of Fe3C/CF; d. EDX analysis of Fe3C/CF) |
对于这种特殊形貌的负载铁的碳纤维形成过程推测如下:一方面,在高温N2气氛处理时,聚丙烯腈(PAN)分子结构通过环化、交联和缩聚等反应在300 ℃左右被氧化时会发生环化而形成含氮杂环结构,在500~600 ℃左右Fe2O3与含氮杂环结构的相互作用形成Fe2.5N(朱珍平等,1997)碳化温度高于700 ℃后,伴随含H、O、N小分子的逸出,Fe2.5 N与含碳物质原位形成新的Fe3C纳米晶,从而形成Fe3C纳米颗粒在碳纤维表面的沉积(Carles et al.,1999);另一方面,由于Fe3C/CF前驱体在热处理过程中会发生分解及聚集形成含铁颗粒,这些分布在纤维内部和表面的含铁颗粒表面被很薄的碳层包覆,这是由于在高温碳化过程中,含铁颗粒除了能够催化促进纳米纤维的碳化外,还能够催化碳材料的再生长,其机理类似于铁物种催化制备碳纳米管或石墨烯等过程(Hou et al.,2004).聚合物碳化时释放的含碳分子遇催化性的Fe3C颗粒被其捕获,在Fe3C颗粒的催化作用下发生二次生长并在铁颗粒表面重新排列,形成Fe3C纳米颗粒被碳层包裹并镶嵌在碳纤维材料表面.
如图 3所示,BET比表面积仪测定了Fe3C/CF催化剂吸脱附曲线及孔径分布变化情况.经测定碳化后的Fe3C/CF催化剂比表面积为343 m2·g-1,吸脱附曲线表现出典型的I型等温线,属于单层或准单层可逆吸附(何余生等,2004).图 3小图中可以看出,Fe3C/CF催化剂的孔径约为0.5 nm,这些微孔可能是含铁颗粒催化纳米纤维在碳化过程中H2、NH3、N2和H2O等小分子气体逸出所致(Lefevre et al.,2000),也可能是Fe3C/CF纳米纤维经N掺杂后可能会有部分活性自由基(—NH—、—H—)的产生,这些自由基与碳载体发生刻蚀反应,导致孔结构的形成(李丽娅等,2003).
 |
| 图 3 Fe3C/CF 催化剂的氮吸附-脱附曲线和孔径分布 Fig. 3 Nitrogen adsorption-desorption isotherms and pore size distribution of Fe3C/CF |
图 4中比较了不同pH值条件下Fe3C/CF非均相电芬顿降解DMA的效果,pH分别为2、3、4和7时,反应360 min,DMA的去除率分别为35%、96%、20%和10%,pH值对于Fe3C/CF非均相催化降解DMA影响显著.一方面pH影响·OH的生成量,决定了电芬顿反应速率;另一方面,适宜的pH促使铁离子的溶出,在阴极实现Fe3+/ Fe2+的原位转化(邱珊等,2014).
 |
| 图 4 不同初始pH对催化的影响(电流强度I=0.15 A,Fe3C/CF催化剂投加剂量500 mg·L-1,氧气通量40~60 mL·min-1,DMA溶液初始浓度C0=5 mg·L-1) Fig. 4 Effect of initial pH on the catalytic oxidation(Current intensity: 0.15 A;Fe3C/CF dosage: 500 mg·L-1;oxygen flux: 40~60 mL·min-1;DMA concentration: 5 mg·L-1) |
当pH值较高时,会抑制Fe3C/CF催化剂反应中Fe3+ /Fe2+的界面转化过程,从而阻碍了·OH 的产生,同时界面上溶出的Fe3+和Fe2+可能水解形成沉淀(Xu et al.,2004).
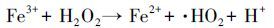
|
(1) |
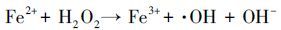
|
(2) |
pH为2,反应60 min时,DMA降解效率下降,这与Fe3C/CF催化剂结构被破坏有关,经分析此时约有40%的铁溶出.因此,在Fe3C/CF非均相电芬顿降解DMA时,初始pH保持在3左右较为合适.
3.2.2 DMA初始浓度对DMA降解的影响图 5中DMA溶液在初始浓度分别为1 mg·L-1、2 mg·L-1、5 mg·L-1和10 mg·L-1,反应360 min,DMA的去除率分别为90%、94%、96%和95%,DMA的去除效率未受DMA初始浓度的影响.足量·OH存在时,DMA初始浓度的增加在某种程度上增大有效碰撞几率,维持了DMA稳定的降解效率.
 |
| 图 5 不同DMA初始浓度对催化的影响(初始pH=3,电流强度I=0.15 A,Fe3C/CF投加剂量500 mg·L-1,氧气通量40~60 mL·min-1) Fig. 5 Effect of initial DMA concentration on the catalytic oxidation(Initial pH: 3;current intensity: 0.15 A;Fe3C/CF dosage:500 mg·L-1;oxygen quantity: 40~60 mL·min-1) |
图 6中电流强度分别为0 A、0.05 A、0.1 A、0.15 A和0.2 A,反应360 min,DMA去除率分别为6%、50%、77%、96%和79%.去除率随电流强度增加有所提升,当电流强度大于0.15 A去除率有所下降.在酸性条件下,伴随电流强度的增强,单位时间补给的电子随之增多,有利于O2得电子在阴极ACF表面转化为H2O2,但是电流强度并不是与降解效率呈正相关: 一方面当电流强度过大会增强极化反应,O2得电子可直接转化为H2O(贺文静等,2013);另一方面 H2O2初始浓度升高,增加了活性羟基自由基的产生,过量产生的活性·OH不仅发生自身猝灭,同时也被H2O2捕获,造成H2O2与·OH相互消耗(Kremer,1999).
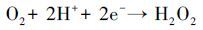
|
(3) |
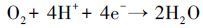
|
(4) |
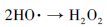
|
(5) |
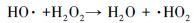
|
(6) |
 |
| 图 6 不同电流强度对催化的影响(初始pH=3;Fe3C/CF投加量500 mg·L-1;氧气通量40~60 mL·min-1;DMA初始浓度C0=5 mg·L-1) Fig. 6 Effect of current strength on the catalytic oxidation(Initial pH: 3;Fe3C/CF dosage:500 mg·L-1;oxygen flux: 40~60 mL·min-1;initial DMA concentration: 5 mg·L-1) |
由图 7可见,无催化剂的电催化反应对DMA几乎无降解作用,说明酸性体系下电产H2O2难以氧化DMA.添加催化剂后,随着增加Fe3C/CF催化剂投量,DMA去除率迅速上升,增加Fe3C/CF催化剂投量为电芬顿提供了足够的反应界面,保证了单位时间·OH产生速率和数量.当投加量达500 mg·L-1时,DMA去除率达到96%,继续增加催化剂投量对DMA降解效率提高并不明显.当Fe3C/CF催化剂投量为1000 mg·L-1时,DMA去除效率反而有所降低,这说明催化剂表面活性位点会在反应过程中捕获起催化作用的·OH及其它中间态活性物质,进而抑制了催化反应过程(Zhang et al.,2008).
 |
| 图 7 不同催化剂投量对催化的影响(初始pH=3,电流强度I=0.15 A,氧气通量40~60 mL·min-1,DMA初始浓度C0=5 mg·L-1) Fig. 7 Effect of catalyst dosage on the catalytic oxidation(Initial pH: 3; current intensity: 0.15 A;oxygen flux: 40~60 mL·min-1;initial DMA concentration: 5 mg·L-1) |
ESR检测发现,纳米Fe3C/CF可作为电芬顿催化剂产生·OH(图 8b),·OH将DMA降解.在不同反应阶段溶液均可检出MMA,但是并未检出无机砷.在反应过程中,纳米Fe3C/CF催化剂慢慢地附着在阴极表面,当反应60 min时,Fe3C/CF催化剂完全附着于阴极表面.取各反应时间的Fe3C/CF催化剂颗粒,置于浓盐酸溶液中超声溶解,检测出As(V),表明DMA的降解产物处理MMA外还有As(V)的生成,部分甲基在·OH作用下也可能转化成CO2和甲醇(Xu et al.,2008).MMA和As(V)的生成情况如图 8所示,反应360 min,DMA浓度为0.29 mg·L-1,MMA浓度为2.9 mg·L-1,As(V)浓度为1.8 mg·L-1,整个反应过程中总砷含量保持平衡(图 8a).
 |
| 图 8 Fe3C/CF催化反应中砷的转化 Fig. 8 Conversion of arsenic in the catalytic reaction over Fe3C/CF |
1) 利用静电纺丝模板煅烧法成功制备出新型纳米Fe3C/CF非均相电芬顿催化剂,催化剂比表面积为343 m2·g-1,孔径约为0.5 nm.
2) 纳米Fe3C/CF催化剂可非均相电芬顿降解DMA,反应360 min,DMA降解效率为96%,其最佳反应条件为pH = 3、DMA初始浓度5 mg·L-1、电流强度0.15 A、催化剂投量500 mg·L-1.
3) DMA通过非均相电芬顿过程被降解为MMA和As(V),降解产物As(V)可被同步吸附在Fe3C/CF催化剂表面.该过程中由·OH起主要氧化作用并且总砷含量始终保持平衡.
| [1] | Braman R S, Foreback C C. 1973. Methylated forms of arsenic in environment[J]. Science , 182 (4118) : 1247–1249. |
| [2] | Brillasa E, Sauleda R, Casado J. 1999. Use of an acidic Fe/O2 cell for wastewater treatment:Degradation of aniline[J]. Journal of the Electrochemical Society , 146 (12) : 4539–4543. DOI:10.1149/1.1392671 |
| [3] | Carles V, Alphonse P, Tailhades P, et al. 1999. Study of thermal decomposition of FeC2O4 center dot 2H2O under hydrogen[J]. Thermochimica Acta , 334 (1/2) : 107–113. |
| [4] | Dopp E, Hartmann L M, Florea A M, et al. 2004. Environmental distribution, analysis, and toxicity of organometal(loid) compounds[J]. Critical Reviews in Toxicology , 34 (3) : 301–333. DOI:10.1080/10408440490270160 |
| [5] | Guivarch E, Trevin S, Lahitte C, et al. 2003. Degradation of azo dyes in water by Electro-Fenton process[J]. Environmental Chemistry Letters , 1 (1) : 38–44. DOI:10.1007/s10311-002-0017-0 |
| [6] | 何余生, 李忠, 奚红霞, 等.2004. 气固吸附等温线的研究进展[J]. 离子交换与吸附 , 2004, 20 (4) : 376–384. |
| [7] | 贺文静, 兰华春, 赵旭, 等.2013. 活性炭纤维电极产生过氧化氢的影响因素与机制研究[J]. 环境科学学报 , 2013, 33 (3) : 725–729. |
| [8] | Hou H, Reneker D H. 2004. Carbon Nanotubers on Carbon Nanofibers: A Novel Structure Based on Electrospun Polymer Nanofibers[J]. Advanced Materials , 16 (1) : 69–73. DOI:10.1002/(ISSN)1521-4095 |
| [9] | Hu C Z, Chen Q X, Liu H J, et al. 2015. Coagulation of methylated arsenic from drinking water: Influence of methyl substitution[J]. Journal of Hazardous Materials , 293 : 97–104. DOI:10.1016/j.jhazmat.2015.03.055 |
| [10] | Jing C Y, Meng X G, Liu S Q, et al. 2005. Surface complexation of organic arsenic on nanocrystalline titanium oxide[J]. Journal of Colloid and Interface Science , 290 (1) : 14–21. DOI:10.1016/j.jcis.2005.04.019 |
| [11] | Kitchin K T. 2001. Recent advances in arsenic carcinogenesis: Modes of action, animal model systems, and methylated arsenic metabolites[J]. Toxicology and Applied Pharmacology , 172 (3) : 249–261. DOI:10.1006/taap.2001.9157 |
| [12] | Kremer M L. 1999. Mechanism of the Fenton reaction[J]. Evidence for a new intermediate[J].Physical Chemistry Chemical Physics , 1 (15) : 3595–3605. DOI:10.1039/a903915e |
| [13] | Lefevre M, Dodelet J P, Bertrand P. 2000. O2 reduction in PEM fuel cells: Activity and active site structural information for catalysts obtained by the pyrolysis at high temperature of Fe precursors[J]. Journal of Physical Chemistry B , 104 (47) : 11238–11247. DOI:10.1021/jp002444n |
| [14] | 李丽娅, 黄启忠, 张红波.2003. PAN基预氧丝炭化过程中的热收缩行为[J]. 炭素 , 2003, 01 : 3–6. |
| [15] | Liu X P, Zhang W F, Hu Y A, et al. 2013. Extraction and detection of organoarsenic feed additives and common arsenic species in environmental matrices by HPLC-ICP-MS[J]. Microchemical Journal , 108 (3) : 38–45. |
| [16] | Nakajima T, Xu Y H, Mori Y, et al. 2005. Combined use of photocatalyst and adsorbent for the removal of inorganic arsenic(Ⅲ) and organoarsenic compounds from aqueous media[J]. Journal of Hazardous Materials , 120 (1/3) : 75–80. |
| [17] | Outran M A, Pinson J. 1995. Hydroxylation by electro-chemically generated ·OH radicals. Mono- and poly hydroxylation of Benzoic Acid: Productsand isomers's distribution[J]. Journal of Physical Chemistry , 99 (38) : 13948–13954. DOI:10.1021/j100038a029 |
| [18] | Petrick J S, Ayala-Fierro F, Cullen W R, et al. 2000. Aposhian HV. Monomethylarsonous acid (MMA(Ⅲ)) is more toxic than arsenite in Chang human hepatocytes[J]. Toxicology and Applied Pharmacology , 163 (2) : 203–207. DOI:10.1006/taap.1999.8872 |
| [19] | 邱珊, 柴一荻, 古振澳, 等.2014. 电芬顿反应原理研究进展[J]. 环境科学与管理 , 2014, 09 : 55–58. |
| [20] | Shimizu M, Ginder-Vogel M, Parikh S J, et al. 2010. Molecular Scale Assessment of Methylarsenic Sorption on Aluminum Oxide[J]. Environmental Science &Technology , 44 (2) : 612–617. |
| [21] | Wang A M, Qu J H, Ru J, et al. 2005. Mineralization of an azo dye Acid Red 14 by electro-Fenton's reagent using an activated carbon fiber cathode[J]. Dyes and Pigments , 65 (3) : 227–233. DOI:10.1016/j.dyepig.2004.07.019 |
| [22] | 奚倩, 赵雅娉, 张警予, 等.2014. 利用电子自旋共振技术研究海参提取液体外抗氧化活性[J]. 食品与机械 , 2014, 05 : 36–40. |
| [23] | 肖华, 周荣丰.2004. 电芬顿法的研究现状与发展[J]. 上海环境科学 , 2004, 23 (6) : 253–256. |
| [24] | Xu X R, Zhao Z Y, Li X Y, et al. 2004. Chemical oxidative degradation of methyl tert-butyl ether in aqueous solution by Fenton's reagent[J]. Chemophere , 55 (1) : 73–79. DOI:10.1016/j.chemosphere.2003.11.017 |
| [25] | Xu Z, Jing C, Li F, et al. 2008. Mechanisms of photocatalytical degradation of monomethylarsonic and dimethylarsinic acids using nanocrystalline titanium dioxide[J]. Environmental Science &Technology , 42 (7) : 2349–2354. |
| [26] | Zhang G Q, Yang F L, Gao M M. 2008. Electro-Fenton degradation of azo dye using polypyrrole/ anthraquinone disulphonate composite film modifiedgraphite cathode in acidic aqueous solutions[J]. Electrochimica Acta , 53 (5) : 5155–5161. |
| [27] | Zhu S, Chen Z, Li B, et al. 2011. Nitrogen-doped carbon nanotubes as air cathode catalysts in zinc-air battery[J]. Electrochimica Acta , 56 (14) : 5080–5084. DOI:10.1016/j.electacta.2011.03.082 |
| [28] | 朱珍平, 顾永达, 吴东, 等.1997. 聚丙烯腈脱氮碳化过程中负载铁的化学形态[J]. 燃料化学学报 , 1997, 25 (2) : 148–151. |