 2016, Vol. 36
2016, Vol. 36



砷是自然界中的一种微量元素,可赋存于多种天然矿物中.在某些地球化学、水文地质或人为因素的诱导下,含水介质中的砷可释放进入地下水中,进而导致水相砷含量异常(Nordstrom,2002).长期饮用高砷地下水可导致一系列的健康问题,包括皮肤癌、肺癌、肝脏和肾脏疾病等(Duker et al.,2005).鉴于此,世界卫生组织(WHO)及我国将饮用水砷标准限定为10 μg·L-1(中国国家标准化管理,2007; WHO,2011).
高砷地下水在世界范围内广泛分布,全球约70多个地区近1.5亿人口均不同程度地受到高砷地下水的威胁,尤其是印度、孟加拉、越南、缅甸、智利、阿根廷、匈牙利、美国和中国(Smedley et al.,2002).我国原生高砷地下水主要分布于山西大同盆地、内蒙古河套平原及新疆、台湾等地区,共约1850万人口受到高砷地下水的威胁,地下水砷含量最高可达约3 mg·L-1(Guo et al.,2014).近年来,针对砷污染地下水原位修复的新技术不断涌现,其主要通过砷与铁氧化物/氢氧化物之间的吸附与共沉淀反应来实现对地下水中的砷去除(Brunsting et al.,2014; Xie et al.,2015).其中,通过向含水层注入氧化剂与铁盐,生成铁氧化物来吸附砷是最为熟知的一类地下水砷原位修复方法.尽管铁氧化物/氢氧化物作为高效的砷去除材料被广泛应用,但这类材料的显著缺陷在于不能修复具有强还原性的高砷地下水.大量研究表明,还原环境中,砷的地球化学循环过程与铁硫化物密切相关(Huerta-Diaz et al.,1992; Morse et al., 1999).众多学者针对铁硫矿物对砷污染水体的修复能力开展的研究(Farquhar et al.,2002; Han et al.,2000; Zouboulis et al.,1993)表明,砷易与硫化物反应形成砷的硫化物,如毒砂、雄黄、雌黄等(Darnley et al.,1995);在还原条件下,铁的硫化物,如陨硫铁矿、四方硫铁矿、黄铁矿等,对砷均有良好的去除效果(Bostick et al.,2003; Wolthers et al.,2005).
尽管铁硫化物作为优良的除砷材料表现出良好的应用前景(Prucek et al.,2013;Prucek et al.,2013;Su et al.,2008 ; Guo et al.,2005; Jang et al.,2007; Gupta et al.,2009),但应用含水层负载铁硫化物开展地下水砷原位修复的研究尚未见报导.鉴于此,本文提出原位铁硫化物含水介质搭载技术,利用FeSO4和Na2S作为联合注入试剂,通过室内模拟,模拟真实地下水环境,探讨搭载介质原位除砷能力及所搭载矿物的稳定性,综合评价该技术在实地处理原生高砷地下水的可操作性.具体研究目标包括:1探讨原位除砷柱实验最佳搭载条件,如最佳Fe/S、最佳负载时间等,以期对场地尺度实验提供技术支持;2通过柱实验模拟原位除砷效果,从而对场地尺度的除砷效果进行预测;3通过除砷材料表征研究对硫化物除砷机制进行初步探讨.
2 材料和方法(Materials and methods) 2.1 材料与试剂本研究中所有实验柱均为长L=30 cm,内径ID=4.5 cm,容积V柱=477 cm3的有机玻璃柱(实验柱符合L≥4×ID,能确保填装柱有均匀的有效孔隙度),实验柱及相关配件(如导管、密封圈等)均用质量分数为15%的稀盐酸浸泡48 h后,用去离子水冲洗洁净,晾干备用.选用石英砂(粒径为0.30~0.84 mm)模拟含水层介质颗粒,用去离子水反复清洗洁净备用.除砷搭载材料采用分析纯FeSO4·7H2O及分析纯Na2S·10H2O.
2.2 制备流程采用湿法填装实验柱,填装过程无氧水充满整个实验柱,以确保石英砂均匀填装且无氧气介入.采用多通道泵和高强度PVC管线调节柱内水流速度.除砷材料搭载采用图 1所示装置及四步循环法完成,具体实验步骤如下:1以νi=4 mL·min-1进水速率泵入5 mmol·L-1 FeSO4溶液(厌氧条件,DO< 0.01 mg·L-1)1 min;2以相同进水速率泵入无氧去离子水1 min;3以相同进水速率继续泵入4 mmol·L-1 Na2S溶液1 min;4不改变进水速率,再次泵入无氧去离子水1 min.重复循环上述4个步骤,至实验柱中石英砂颜色无明显变化且出水中Fe含量保持恒定时,除砷材料制备完成.
 |
| 图 1 除砷柱制备示意图 Fig. 1 Schematic figure of the experimental column set-up |
选定FeSO4和Na2S溶液浓度分别为5 mmol·L-1和4 mmol·L-1,观察不同负载时间内实验柱颜色的变化,确定最佳负载时间,该实验柱标为柱A.制备过程可能发生如下反应( Hunger et al.,2007):
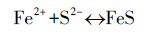
|
(1) |
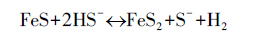
|
(2) |
按上述方法准备3个实验柱,将FeSO4与Na2S溶液浓度分别设定为1与0.8 mmol·L-1、2与1.6 mmol·L-1、4与3.2 mmol·L-1,依2.2节所述方法制备除砷材料,观察同时段内不同实验柱颜色变化,寻找最佳注入液浓度配比.该组实验柱依次标为柱B-1、柱B-2、柱B-3.
2.4 除砷实验 2.4.1 荧光素钠穿透试验选取荧光素钠为惰性示踪剂,监测其穿透过程.先泵入10V孔体积的无氧去离子水冲洗实验柱A,再以注入速率为νj= 4 mL·min-1向实验柱A中连续泵入浓度89.4 mg·L-1的荧光素钠溶液.泵入过程中每60 s采集1件出水样品,并立即测定荧光素钠浓度,绘制荧光素钠穿透曲线.
2.4.2 砷(As(Ⅲ))穿透试验在还原环境下,地下水中的砷主要以As(Ⅲ)形式存在( Inskeep et al.,2002; Nordstrom et al.,2003; Xieet al.,2008),因此,本实验重点探讨了As(Ⅲ)的穿透行为.
通过对比荧光素钠溶液与As(Ⅲ)溶液的穿透曲线,评价该材料的除砷效率,通过检测搭载Fe含量和吸附As总量,计算除砷容量.具体实验过程如下.
第一步:以νj=4 mL·min-1的速率连续泵入10V孔的无氧去离子水冲洗柱A.
第二步:不改变进水速率,将1000 μg·L-1 NaAsO2(以As浓度计)溶液泵入实验柱A中,每隔1 h收集一份出水样品(10 mL),取其中5 mL进行砷形态分离(分离步骤见2.6.1节),剩余样品滴加1滴优级纯浓HCl酸化至pH<2,避光冷藏保存,在72 h内测定As浓度.同时,另取约20 mL流出液至50 mL PET瓶中,采用HACH便携式水质分析仪测试其pH、Eh、EC值.
第三步:当流出液与注入液中As浓度一致并在一段时间内保持稳定时,终止实验,将此除砷处理后的实验柱标记为柱C,避光保存待用.
将实验柱C中的负载石英砂全部取出,先加入500 mL 6 mol·L-1 HCl溶液,室温振荡提取30 min,静置12 h后,收集溶液,并再次用6 mol·L-1 HCl重复上述清洗过程,直到石英砂表面无黑褐色残留为止,最后用去离子水清洗石英砂3遍,测定收集的溶液中铁和砷的含量,计算除砷容量.
2.5 原位除砷模拟依照2.1节所述方法准备空白石英砂柱.为模拟实际地下水环境的原位处理过程,同时弱化其他离子影响,在严格厌氧条件下,以1000 μg·L-1 NaAsO2溶液作为污染物质模拟原位除砷过程.采用4步交替循环法,以νi=4 mL·min-1的进水速率向空白砂柱中交替注入5 mmol·L-1 FeSO4溶液、1000 μg·L-1 NaAsO2溶液、Na2S溶液(H2S饱和溶液中加入Na2S,S2-浓度为4 mmol·L-1)及1000 μg·L-1 NaAsO2溶液各持续1 min.每4 h收集一份流出液样品,加入优级纯HCl调节pH至2,冷藏保存待测,约800 h后完成原位除砷模拟实验.样品采集的同时,取约20 mL样品至50 mL PET瓶中,采用HACH便携式水质分析仪测试其pH、Eh、EC值.该实验柱标为D.
实验完成后,收集柱内负载石英砂,按2.3节处理方法测定铁和砷的含量,计算砷的吸附量.
2.6 分析方法 2.6.1 砷形态分离实验柱出水样品立即用砷形态分离小柱分离As(Ⅲ)与As(V),方法如下:1用注射器吸取10 mL 体积分数50%(1∶1)的甲醇活化液,接口依次接上一次性针头过滤器和砷形态分离小柱,以1~2滴·s-1的速度慢慢推出将柱子活化,直至将甲醇溶液排挤干净;2准确移取5 mL水样以1~2滴·s-1的 速率分离样品砷形态,出水口所接样品为As(Ⅲ),滴加1~2滴浓盐酸至pH为2,样品于4 ℃避光保存;同样保证柱中溶液排挤干净;3准确吸取5 mL 0.5 mol·L-1盐酸洗脱分离柱,以1~2滴·s-1的速度流出,此部分为As(V),样品于4 ℃避光保存;4使用后的分离小柱再经10 mL 0.5 mol·L-1盐酸和10 mL去离子水各清洗2遍,重复上述过程完成所有样品分离工作.
2.6.2 样品分析样品收集完成后,用便携式分光光度计(HACH,DR2800)测定荧光素钠浓度;用原子荧光光谱仪(AFS,北京海天仪器)测定砷含量;用还原剂(10%盐酸羟胺)还原提取液中的Fe至Fe(Ⅱ),后用便携式分光光度计(HACH,DR2800)邻菲罗啉法测定铁含量.
取实验柱A、C和D中的负载石英砂各10 g,室温晾干后避光隔绝空气保存.选取负载石英砂颗粒,经喷金处理后,采用超高分辨率场发射扫描电子显微镜(日立,SU8010)观察石英砂表面铁搭载的微观形貌,并采用配套的X射线能量色散谱仪表征目标微区的化学组成和元素含量.此外,取平行样品5 g,与5 g优级纯KBr固体混匀,用玛瑙研钵研磨至100目后,以KBr粉末为背景值,采用傅里叶变换-红外光谱仪(赛默-飞世尔,Nicolet6700)分析矿物表面砷与铁的结合形态.
当实验柱完全充水后,使柱中水在重力作用下全部释出,用天平测定出水质量m孔,作为柱内孔隙水质量,取多次实验测定值的平均值,计算柱孔隙体积V孔,从而得到石英砂柱孔隙度η(式(3)).
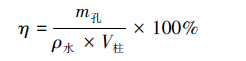
|
(3) |
定义无因次阻滞因子(R)用于表征柱中砷相对于惰性示踪剂的延迟穿透(van Halem et al.,2010).R值计算公式如下:
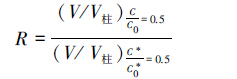
|
(4) |
式中,C为出水中As的测量值(μg·L-1),C0为注入As溶液初始值(μg·L-1),惰性示踪剂为荧光素钠,C*为出水中荧光素钠的测量值(mg·L-1),C*0为注入荧光素钠溶液初始值(mg·L-1).V/V柱值为出水的体积(V)与柱孔隙体积(V柱)的比值.
3 结果与讨论(Results and discussion) 3.1 条件优化原位除砷过程中,除砷材料制备时间(即试剂负载时间)、FeSO4与Na2S溶液注入浓度及实验前后含水层(即实验柱)孔隙度的变化均可不同程度地影响地下水的修复效果(Han et al.,2011).本实验通过改变室内柱实验条件,探讨除砷材料制备的最佳负载时间、溶液最佳注入浓度及相应的孔隙度变化.通过条件优化可以大量节省注入时间及试剂,并且对后期原位除砷改水实践提供技术支持.
3.1.1 最佳负载时间空白实验柱A在搭载过程中石英砂表面的灰黑色随时间推移逐渐加深.当负载时间t达120 h后,实验柱中石英砂涂层颜色无明显变化,出水中Fe含量也无明显变化,因此,将120 h设定为最佳负载时间.
除砷材料制备完成后,实验柱中石英砂表面呈均匀灰黑色,推测该灰黑色物质可能为铁的(多)硫化物,即FeSm.由于无氧去离子水的交替注入形成反应缓冲区,该实验过程中无堵塞现象发生.
3.1.2 FeSO4与Na2S最佳注入浓度FeSO4与Na2S溶液浓度过高,可能会导致柱体堵塞,浓度过低则会延长材料制备时间且会影响处理效果,因此,选取最佳的注入浓度对后续试验结果和后期场地原位修复的应用至关重要.
当FeSO4与Na2S溶液浓度较低(实验柱B-1、B-2)时,制得的实验柱下端石英砂表面颜色偏浅且被氧化,即使增加负载时间,石英砂柱表面颜色变化仍不明显,说明注入浓度偏低时石英砂表面无法负载足量的铁硫化物沉淀,其原因可能是在水流的冲刷作用下铁硫化物的附着量小于冲刷量.当试剂浓度增大到4 mmol·L-1和3.2 mmol·L-1时,可观察到在48 h后试验柱颜色反而变为浅黄色,可能是由于在生成铁硫化物的同时铁硫化物被氧化.当FeSO4与Na2S溶液浓度分别增加至5 mmol·L-1与4 mmol·L-1时,实验柱颜色呈现较为均匀的黑褐色,说明铁硫化物的负载量较多,分布相对均匀,有利于除砷效果的提高.因此,本研究选定FeSO4与Na2S溶液的最佳注入浓度分别为5 mmol·L-1与4 mmol·L-1.
本实验测得,上述3种不同注入液浓度条件下制得的负载石英砂柱(实验柱B-1、B-2、B-3)的孔隙度变化不明显(分别为22.31%、22.30%、22.35%),说明原位搭载试验完成不会阻塞含水层.因此,将孔隙度值22.31%近似作为上述不同条件下制得实验柱的孔隙度.
3.2 搭载材料除砷 3.2.1 除砷效果图 2a为负载石英砂柱(柱A)荧光素钠穿透曲线,初始浓度为89.4 mg·L-1的荧光素钠溶液以4 mL·min-1的速率注入实验柱后,从图中可以看出,荧光素钠在25 min(100 mL,0.94V孔)时开始穿透,完全穿透时间约为1.25 h.
 |
| 图 2 荧光素钠(a)、NaAsO2(b)穿透曲线及出水中As(Ⅲ)与总砷的比率(c) Fig. 2 The breakthrough curve for fluorescein sodium(a)NaAsO2(b)and the ratio of As(Ⅲ)in the effluents(injection of NaAsO2)(c) |
初始浓度为1000 μg·L-1的NaAsO2溶液穿透曲线如图 2b所示,As(Ⅲ)在约1 h(240 mL,2.26 V孔)时开始穿透,在100 h(24 L,225.52 V孔)时趋于稳定.As(Ⅲ)的穿透明显滞后,其阻滞因子R约为37,当As(Ⅲ)通过铁硫化物试验柱时,As(Ⅲ)被吸附,从而造成砷穿透的严重滞后.在完全穿透后对矿物进行提取测试分析,结果显示,As(Ⅲ)柱内搭载量为0.91 g,吸附砷总量为40.91 mg,即As(Ⅲ)在铁硫化物上的动态吸附平衡量为44.94 mg·g-1(0.034 mol·mol-1)(以每 g或mol Fe上吸附的As(Ⅲ)量(mg,mol)计).
对实验柱出水样品进行砷形态分离,柱A(注入液为As(Ⅲ))出水中As(Ⅲ)与出水中总As比率结果如图 2c所示,可观察到在初始阶段(0~30 h)出水中As(Ⅲ)比率呈快速上升趋势,As(Ⅲ)最高含量可与总砷含量相当,在经过一段急速增长阶段后(30 h后)As(Ⅲ)比率有所下降并趋于稳定,最后As(Ⅲ)比率稳定于0.7~0.8之间.
Bostick与Fendorf(2003)的研究表明,As(Ⅲ)与铁硫化物可发生如下反应过程:
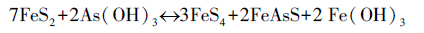
|
(5) |
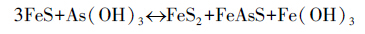
|
(6) |
As(Ⅲ)与铁硫化物反应生成Fe(OH)3,而Fe(OH)3对As具有明显的去除效果.当环境中存在Fe(OH)3时,会将As(Ⅲ)氧化为As(V)(Amstaetter et al.,2009).整个实验环境处于还原条件,不易于As(Ⅲ)的氧化;另外,As(Ⅲ)在水溶液中基本不带电荷,不利于砷在Fe(OH)3表面的吸附.因此,可观察到当As(Ⅲ)注入实验柱后,出水中As(Ⅲ)/总As值较高,其变化区间为0.7~0.8.
3.2.2 除砷材料解析对除砷材料进行扫描电镜及EDS能谱分析(图 3a,3b由扫描电镜图可看出,除砷前,负载石英砂表面负载一层结晶程度较好、呈短柱状及团簇状矿物,由其EDS能谱图可看出,铁、硫含量较高,且Fe/S比约为1/2,推测形成的矿物可能为黄铁矿、无定型态的铁硫化物.
 |
| 图 3 除砷前扫描电镜及其EDS图(a,b)和As(Ⅲ)反应后扫描电镜及其EDS图(c,d) Fig. 3 Scanning electron microscopy(SEM)image and energy dispersive X-rayspectrum(EDS)of Fe-coating quartz sand before As loading(a,b),after As(Ⅲ)loading(c,d) |
当As(Ⅲ)吸附在负载矿物表面并达到动态吸附平衡后,其扫描电镜结果如图 3b所示,砷吸附后矿物表面形态发生明显变化,由反应前的短柱状、团簇状变为反应后的片状堆积,且结晶程度变差,由此推断可能发生除表面吸附反应外的其他反应过程.由EDS结果(图 3b,3d)可知,Fe/S由吸附前的1/2变为吸附后的1/1,说明As(Ⅲ)在与铁硫化物发生反应时,将S替换,从而形成Fe-As-S类新物质使得固相矿物中S含量降低.
对比吸附As(Ⅲ)前后矿物EDS能谱图发现,砷含量明显增加,从吸附前低于检出限增加到吸附后的3.05%(质量分数),说明所搭载除砷材料对As(Ⅲ)具有显著的去除作用.由3.2.1节分析可知,在As(Ⅲ)与铁硫化物反应时会伴随有氧化还原反应(As(Ⅲ)被Fe(OH)3氧化)发生.
3.3 地下水原位除砷模拟 3.3.1 除砷效果将铁、As(Ⅲ)、硫同时注入实验柱模拟高砷地下水铁硫化物原位处理过程,监测结果如图 4所示.实验柱D出水砷含量开始较大(约730 μg·L-1),随后急剧降低(12 h降至约240 μg·L-1),继而维持在较低水平(0~10 μg·L-1),表明在模拟地下水原位除砷过程中,铁与硫的注入可以有效去除地下水中的砷.在此过程中,由于Fe(Ⅱ)、As(Ⅲ)和S2-同时注入,水相中的砷可能通过吸附或者共沉淀作用被去除(Tokoro et al.,2009).
 |
| 图 4 模拟高砷地下水砷穿透曲线 Fig. 4 The breakthrough curve for simulation of high arsenic groundwater |
实验柱D中铁搭载总量为2.64 g,吸附的砷总量为291.11 mg,吸附能力为0.083 mol·mol-1,大于铁硫化物对As(Ⅲ)的动态吸附量(0.034 mol·mol-1).同时注意到,本实验中砷的穿透更加滞后,砷的吸附远未达到饱和.这对于实际的场地实施是一个重要的发现,因为此除砷材料在运用到高砷含水层时,可能表现出更好的除砷效果(出水中砷的含量保持在低水平)和更长的使用寿命(出水中砷的穿透大大延迟).
3.3.2 除砷机制图 5a为同时注入FeSO4、As(Ⅲ)、Na2S模拟地下水原位除砷后负载矿物的扫描电镜图,砷吸附后矿物结晶程度较好,呈草莓状;由其EDS能谱图(图 5b)可看到,Fe/S约为1/2,砷含量为3.66%(质量分数),据此推测为含砷草莓状黄铁矿( Neumann et al.,2013).在还原条件下,As(Ⅲ)会替换FeS2中S( Blanchard et al.,2007),因此,形成的砷黄铁矿中Fe/S会略高于1/2.红外光谱(图 6)分析表明,As(Ⅲ)在铁硫化物(FeS和Fe2S)表面的吸附较为复杂,并且与pH直接相关.在低pH条件下,As(Ⅲ)的吸附遵循表面的配体交换作用和伴随质子的消耗(式(7)).在高的pH条件下,吸附作用涉及的是强的内层络合物的形成,区别于表面的羟基交换作用(式(8)).此外,高浓度硫化物存在时,会促使砷硫化物的形成(式(9)).理想条件下,As(Ⅲ)还可以与FeS反应生成类似于FeAsS的沉淀,促使砷更稳定地进入矿物相(式(10)).
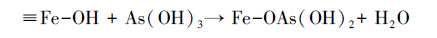
|
(7) |

|
(8) |
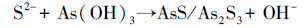
|
(9) |
|
|
(10) |
 |
| 图 5 同时注入Fe、As、S扫描电镜及其EDS图 Fig. 5 Scanning electron microscopy(SEM)image and energy dispersive X-rayspectrum(EDS)of joint injection of Fe、As、S |
 |
| 图 6 实验柱A、实验柱C红外结果 Fig. 6 The infrared spectroscopy results for column A and column C |
对比不同条件下红外分析结果可知:在只有H2S存在时,小波数处与背景基本重合.1695 cm-1和1882 cm-1处出现小的吸收峰,应该是As-OH键的ν(O-H)振动吸收峰.同时,在3104 cm-1处的羟基吸收峰较宽,说明反应后的-(O-H)的键合作用相对宽松.据此反应可能按照式(7)进行.此时处于低pH条件下,实验过程观测到黑色无定型沉淀的产生,推测是As在新生成的FeS无定型沉淀固相表面的羟基交换作用.相比而言,在H2S和Na2S存在时,小波数处(图中右边5个峰)的吸收峰强明显减弱.注意到,与只有H2S时相比,1695 cm-1稍蓝移至1734 cm-1处,而3104 cm-1处红移至3068 cm-1处,并且大波数处各峰吸收明显增强.这指示了不同于前者的砷结合方式,推测是式(8)的作用机制.459 cm-1和692cm-1等处吸收峰的减弱是由于ν(As-OH)振动被与之结合的多硫结构所约束,化学键合作用增强,因此,吸收减少.3104 cm-1处红移至3068 cm-1处及峰强的增加可能是由于与砷结合的羟基所受的束缚减弱,受到了与砷结合的多硫结构的负电性的影响.
4 结论(Conclusions)1) 采用四步交替循环法,交替注入5 mmol·L-1 FeSO4、无氧去离子水和4 mmol·L-1 Na2S,注入时间为120 h时,可在石英砂表面形成均匀的铁硫化物搭载,且不会造成堵塞.
2) NaAsO2在负载石英砂柱中的完全穿透时间(约100 h)远大于荧光素钠(约1.25 h),砷的阻滞因子达37,砷动态吸附容量为0.034 mol·mol-1.在反应过程中会伴随氧化还原反应发生.
3) 通过同时注入FeSO4、NaAsO2和Na2S模拟含水层原位除砷发现,当地下水砷含量为1000 μg·L-1时,材料制备过程中水相砷处理效果很好,随着时间延长As(Ⅲ)会被全部去除,砷吸附量为0.083 mol·mol-1.砷的固定是一个吸附-共沉淀的过程.
| [1] | Amstaetter K, Borch T, Larese-Casanova P, et al. 2009.Redox transformation of arsenic by Fe(Ⅱ)-activated goethite (α-FeOOH)[J]. Environmental Science&Technology, 44 (1): 102–108. |
| [2] | Blanchard M, Alfredsson M, Brodholt J, et al. 2007.Arsenic incorporation into FeS2 pyrite and its influence on dissolution: a DFT study[J]. Geochimica et Cosmochimica Acta, 71 (3): 624–630. |
| [3] | Bostick B C, Fendorf S. 2003.Arsenite sorption on troilite (FeS) and pyrite (FeS2)[J]. Geochimica et Cosmochimica Acta, 67 (5): 909–921. |
| [4] | Brunsting J H, McBean E A. 2014.In situ treatment of arsenic-contaminated groundwater by air sparging[J]. Journal of Contaminant Hydrology, 159 : 20–35. |
| [5] | Darnley A G,Björklund A,Bolviken B,et al.1995.A Global geochemical database: Recommendations for international geochemical mapping[R].Final Report of IGCP Project 259.Paris: UNESCO |
| [6] | Duker A A, Carranza E J M, Hale M. 2005.Arsenic geochemistry and health[J]. Environment International, 31 (5): 631–641. |
| [7] | Farquhar M L, Charnock J M, Livens F R, et al. 2002.Mechanisms of arsenic uptake from aqueous solution by interaction with goethite,lepidocrocite,mackinawite,and pyrite: An X-ray absorption spectroscopy study[J]. Environmental Science&Technology, 36 (8): 1757–1762. |
| [8] | Guo H M, Wen D G, Liu Z Y, et al. 2014.A review of high arsenic groundwater in Mainland and Taiwan,China: Distribution,characteristics and geochemical processes[J]. Applied Geochemistry, 41 : 196–217. |
| [9] | Guo W, Hou Y L, Wang S G, et al. 2005.Effect of silicate on the growth and arsenate uptake by rice (Oryza sativa L ) seedlings in solution culture[J]. .Plant and Soil, 272 (1/2): 173–181. |
| [10] | Gupta A, Chauhan V S, Sankararamakrishnan N. 2009.Preparation and evaluation of iron-chitosan composites for removal of As (Ⅲ) and As (V) from arsenic contaminated real life groundwater[J]. Water Research, 43 (15): 3862–3870. |
| [11] | Han J T, Fyfe W S. 2000.Arsenic removal from water by iron-sulphide minerals[J]. Chinese Science Bulletin, 45 (15): 1430–1434. |
| [12] | Han Y S, Gallegos T J, Demond A H, et al. 2011.FeS-coated sand for removal of arsenic(Ⅲ) under anaerobic conditions in permeable reactive barriers[J]. Water Research, 45 (2): 593–604. |
| [13] | Huerta-Diaz M A, Morse J W. 1992.Pyritization of trace metals in anoxic marine sediments[J]. Geochimica et Cosmochimica Acta, 56 (7): 2681–2702. |
| [14] | Hunger S, Benning L G. 2007.Greigite: A true intermediate on the polysulfide pathway to pyrite[J]. Geochemical Transactions, 8 (1): 1. |
| [15] | Inskeep W P,McDermott T R,Fendorf S.2002.Arsenic (V)/(Ⅲ) cycling in soils and natural waters: Chemical and microbiological processes//Frankenberger W T Jr.Environmental Chemistry of Arsenic[M].New York: Marcel Dekker.183-215 |
| [16] | Jang M, Hwang J S, Choi S I. 2007.Sequential soil washing techniques using hydrochloric acid and sodium hydroxide for remediating arsenic-contaminated soils in abandoned iron-ore mines[J]. Chemosphere, 66 (1): 8–17. |
| [17] | Morse J W, Luther Ⅲ G W. 1999.Chemical influences on trace metal-sulfide interactions in anoxic sediments[J]. Geochimica et Cosmochimica Acta, 63 (19/20): 3373–3378. |
| [18] | Neumann T, Scholz F, Kramar U, et al. 2013.Arsenic in framboidal pyrite from recent sediments of a shallow water lagoon of the Baltic Sea[J]. Sedimentology, 60 (6): 1389–1404. |
| [19] | Nordstrom D K. 2002.Worldwide occurrences of arsenic in ground water[J]. Science, 296 (5576): 2143–2145. |
| [20] | Nordstrom D K,Archer D G.2003.Arsenic thermodynamic data and environmental geochemistry[M]//Arsenic in ground water.Springer US,1-25 |
| [21] | Prucek R, Tućek J, Kolar'ík J, et al. 2013.Ferrate (VI)-induced arsenite and arsenate removal by in situ structural incorporation into magnetic iron (Ⅲ) oxide nanoparticles[J]. Environmental Science&Technology, 47 (7): 3283–3292. |
| [22] | Smedley P L, Kinniburgh D G. 2002.A review of the source,behaviour and distribution of arsenic in natural waters[J]. Applied Geochemistry, 17 (5): 517–568. |
| [23] | Su Y H, McGrath S P, Zhu Y G, et al. 2008.Highly efficient xylem transport of arsenite in the arsenic hyperaccumulator Pteris vittata[J]. New Phytologist, 180 (2): 434–441. |
| [24] | Tokoro C, Yatsugi Y, Koga H, et al. 2009.Sorption mechanisms of arsenate during coprecipitation with ferrihydrite in aqueous solution[J]. Environmental Science&Technology, 44 (2): 638–643. |
| [25] | van Halem D, Heijman S G J, Johnston R, et al. 2010.Subsurface iron and arsenic removal: low-cost technology for community-based water supply in Bangladesh[J]. Water Science&Technology, 62 (11): 2702–2709. |
| [26] | W HO. 2011. Guidelines for Drinking-Water Quality (4th ed). Guidelines for Drinking-Water Quality[M]. Geneva: WHO . |
| [27] | Wolthers M, Charlet L, van Der Weijden C H, et al. 2005.Arsenic mobility in the ambient sulfidic environment: Sorption of arsenic (V) and arsenic (Ⅲ) onto disordered mackinawite[J]. Geochimica et Cosmochimica Acta, 69 (14): 3483–3492. |
| [28] | Xie X J,Ellis A,Wang Y.2008.Hydrogeochemistry and sulfur isotopes of high arsenic groundwater of Datong basin,China[A]//American Geophysical Union Fall Meeting Abstracts[C].New York: American Geophysical Union. |
| [29] | Xie X, Wang Y, Pi K, et al. 2015.In situ treatment of arsenic contaminated groundwater by aquifer iron coating: Experimental study[J]. Science of the Total Environment, 527 : 38–46. |
| [30] | Zhang G, Ren Z, Zhang X, et al. 2013.Nanostructured iron (Ⅲ)-copper (Ⅱ) binary oxide: a novel adsorbent for enhanced arsenic removal from aqueous solutions[J]. Water Research, 47 (12): 4022–4031. |
| [31] | Zouboulis A I, Kydros K A, Matis K A. 1993.Arsenic (Ⅲ) and arsenic (V) removal from solutions by pyrite fines[J]. Separation Science and Technology, 28 (15/16): 2449–2463. |
| [32] | 中国国家标准化管理委员会.2007.GB5749-2006 生活饮用水卫生标准[S].北京: 中国标准出版社 |