 2015, Vol. 35
2015, Vol. 35



2. 大连理工大学静电与特种电源研究所, 大连 116024;
3. 丰桥技术科学大学环境与生命科学系, 丰桥441-8580, 日本
2. Institute of Electrostatics and Special Power, Dalian University of Technology, Dalian 116024;
3. Department Environmental and Life Sciences, Toyohashi University of Technology, Toyohashi 441-8580, Japan
氮氧化物(NOx)是燃煤烟气中的主要污染物之一,可诱发酸雨、破坏臭氧层、形成光化学烟雾等危害,进而威胁人类健康和生态环境安全.选择性催化还原(SCR)脱硝技术具有高效、实用性等优点,是当前研究最多、技术最为成熟的NOx末端处理工艺(Lei et al.,2013;Goo et al.,2007;Cheng et al.,2010;Tronconi et al.,2007).SCR技术的高效运行是在合适的温度窗口内,混有还原剂(NH3)的烟气经过SCR脱硝反应器时,在催化剂的作用下,NO被还原成N2(反应(1)).催化剂中毒会严重影响SCR脱硝技术的正常运行,而积灰污染是导致催化剂中毒最复杂、影响最大的一个因素(张烨等,2011).因此,如果能够将SCR脱硝系统置于除尘装置后,消除或降低积灰污染,应可有效延长催化剂的使用寿命.将SCR脱硝系统置于除尘器之后,烟气到达SCR反应器的温度在150 ℃左右,而商用催化剂的最佳工作温度一般为300~400 ℃,在此温度窗口下,单独依靠目前的商用催化剂很难实现NOx的高效脱除(唐晓龙等,2005).
| 4NH3+ 4NO+O2→4N2+6H2O | (1) |
2000年,Koebel等(2000)提出快速SCR脱硝理论,即将部分NO氧化为NO2(反应(2)),当NO与NO2比例达到1时,SCR系统在低温区的脱硝效率会大幅度提升(反应(3)).
| NO+O(O2,O3,…)→NO2 | (2) |
| 2NH3+NO+NO2→2N2+3H2O | (3) |
在含氧氛围里,放电等离子体能够生产多种高氧化电位的氧活性物质,其引发的氧化反应的速率远快于常规的氧化反应(林赫,2002).Kim等(2001)利用脉冲放电协同SCR实现了低温区高效脱硝.但Obradovic等(2011)认为,当直接利用低温等离子体的氧活性物质氧化NO时,烟气长时间与放电电极直接接触,会引起电极腐蚀导致放电不稳定.
针对上述问题,本文提出采用沿面放电氧活性物质注入方法预氧化烟气中的NO,实现快速SCR反应条件,提高低温区SCR的脱硝性能.同时,考察氧活性物质注入对烟气的预氧化过程、氧活性物质注入协同SCR脱硝过程,以及烟气中存在SO2、H2O时,氧活性物质注入降低SO2、H2O对SCR脱硝效率影响的效果.
2 实验装置(Experimental setup)图 1是氧活性物质注入协同SCR脱硝系统的流程图,整个系统主要由配气系统、沿面放电反应器(Surface Discharge Reactor,SDR)、SCR反应装置和气体采样分析系统4部分组成.
 |
| 图 1 氧活性物质注入协同SCR示意图 Figure.1 Schematic diagram of SCR coupled with reactive oxygen species injection |
放电反应器为沿面介质阻挡放电结构,其中,壁厚为2 mm的石英玻璃管作为绝缘介质,玻璃管内壁紧贴线径为0.5 mm、螺距为2 mm的螺旋金属作为高压电极,玻璃管外壁包裹120 mm长的铝箔纸作为接地电极(铝箔纸长度控制放电长度).SCR反应装置中填充商用蜂窝状V2O5-WO3/TiO2催化剂,利用管式炉加热、控温.
低温等离子反应器插进模拟烟道中,清洁空气(注入空气量为75 mL · min-1,占总气量的5%)在放电反应器内被激发形成氧活性物质后进入模拟烟道中,将烟气中的NO部分氧化成NO2,随后进入SCR反应器,完成SCR脱硝过程.模拟烟道中的烟气流量保持在1.5 L · min-1,主要成分为N2、O2、NO、SO2和H2O(气态),其中,NO浓度为220 ppm.模拟烟气中的NO、SO2与NO2浓度采用烟气分析仪(Testo 350)在线监测,NO2、NO、SO2的测试分辨率分别是0.1、1和1 ppm.烟气温度采用热电偶测试,烟气湿度采用温湿度计(HP 21,Rotronic)测试.放电过程中生成的O3的浓度用碘量法滴定.
沿面放电反应器的供电电源采用9 kHz的交流电源,放电峰值电压通过电压探头(P6015A,泰克)与示波器(TDS 2014,泰克)检测,通过V-Q Lissajous图形方法计算放电功率; 放电过程中的能量密度(Special energy density,SED,J · L-1)则通过如下公式计算:
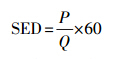
式中,P为沿面放电反应器的放电功率(W),Q为注入气量(L · min-1).
3 结果与讨论(Results and discussion) 3.1 NO 氧化与电压的关系图 2为不同峰值电压下,氧活性物质注入氧化NO的平衡曲线.随着电压的升高,NO浓度几乎呈线性下降,NO2浓度则几乎线性上升,NOx总量基本保持稳定.在此温度条件下,外加电压为2.7 kV时,烟气中NO与NO2的比例为1∶ 1,达到快速SCR的最佳反应条件.
 |
| 图 2 活性氧注入时烟气中NO氧化的平衡曲线(NOx 220 ppm,O2 6%,烟气流速1.5 L · min-1,烟气温度150 ℃,注入空气量75 mL · min-1) Figure.2 Equilibrium curve of NO oxidation by ROS injection |
沿面放电注入反应器内的能量密度、放电生成O3的浓度与放电电压的关系如图 3所示.在外加峰值电压为2 kV时,高压电极与接地极之间开始形成微放电.对于介质阻挡放电结构,在微放电形成的瞬间,放电间隙两端的电压迅速下降甚至放电微通道关闭(王新新,2009),因此,微放电通道中电流的自由增长能够被有效遏制.在微放电通道中,高能电子与气体分子发生非弹性碰撞,几乎将从外加电场中获得的全部能量传递出去(谢维杰等,2008),在此过程中,产生包括O3在内的多种氧活性物质.微放电通道中大于8.4 eV的电子能够通过与氧分子碰撞可以将其分解(反应(4)、(5))(白希尧等,2000),这是形成臭氧(反应(6))至关重要的前提.随着电压增大,越来越多的微放电通道随机的分布在石英管上,注入能量密度也随之提高;电场随着外加电压的增加而增强,电子获得的能量随之增加,高能电子(>8.4 eV)的密度得到进一步的提升,臭氧产量也随之增多.
 |
| 图 3 电压与能量密度及O3浓度的关系(O2 6%,烟气流速1.5 L · min-1,烟气温度150 ℃,注入空气量流速75 mL · min-1) Figure.3 Effect of voltage on the SED and O3 |
| O2+e-→2O(3P)+e- | (4) |
| O2+e→O(3P)+O(1D)+e- | (5) |
| O2+O+M→O3 | (6) |
不同温度下,标准SCR与氧活性物质注入协同SCR的脱硝效率如图 4所示.从图中可以看出,当反应温度低于300 ℃时,氧活性物质注入协同SCR比标准SCR脱硝具有明显地优势.在150 ℃时,氧活性物质注入协同SCR的脱硝效率为51.9%,而标准SCR脱硝效率仅为28.9%,提高了23%.
 |
| 图 4 SCR脱硝与氧活性物质注入协同SCR脱硝的比较(NOx 220 ppm,O2 6%,烟气流速1.5 L · min-1,电压2.7 kV,空速27200 h-1) Figure.4 Comparison between SCR and SCR coupled with reactive oxygen species injection for NOx removal |
氧活性物质注入协同SCR脱除NOx的机理如图 5所示.以O3为主的氧活性物质可将烟气中的NO部分氧化(Wang et al.,2011),当烟气中NO/NO2浓度比为1时,NOx在V2O5-WO3/TiO2催化剂表面发生快速SCR反应.
 |
| 图 5 氧活性物质注入协同SCR催化机理 Figure.5 Coupling mechanism of SCR with reactive oxygen species injection |
在快速SCR过程中,催化剂提供大量的活性点位,吸附的NH3将NO选择性还原为N2,同时,V5+ O被还原反应为V4+—OH(反应(7)).Topsoe等(1995)认为,只有在高温窗口(大于300 ℃)条件下,吸附在催化剂上的O2才能够高效将还原态的V4+—OH再氧化(反应(8)).
| NH3+NO+V5+ O→N2+H2O+V4+—OH | (7) |
| V4+—OH+(1/4)O2→V5+ O+H2O | (8) |
而在快速SCR过程中,V4+—OH的再氧化不仅可以利用催化剂吸附的O2,还能借助气氛中的NO2.特别是在250 ℃以下的低温窗口,与O2相比,化学吸附在催化剂上的NO2氧活性更强.NO2吸附在催化剂表面后,在不可还原氧化位上(S O)转化为NO-3和NO-2(反应(9)).NO-3能够将V4+—OH快速再氧化(反应(10)),而NO-2能够与NH3发生选择性还原反应而转化成N2(反应(11))(Tronconi et al.,2007).在此过程中,V4+—OH的再氧化不再是SCR过程的速率控制步骤,因此,这对提高300 ℃以下的SCR反应速率有重要作用.
| 2NO2+O2-NO-2+NO-3 | (9) |
| V4+—OH+NO-3+H+→V5+ O+NO2+H2O | (10) |
| NH3+NO-2→N2+H2O+O2-+H+ | (11) |
图 6所示为不同温度条件下,SO2对SCR脱硝的影响.从图 6可以看出,在考察的温度范围内,当温度高于150 ℃时,氧活性物质注入可以降低SO2对SCR脱硝效率的影响.以150 ℃下的实验为例,不存在SO2时,标准SCR脱硝效率为28.9%;存在200 ppm的SO2时,标准SCR脱硝效率则为15.7%,下降了13.2%.采用氧活性物质注入协同SCR脱硝,不存在SO2时的脱硝率为51.9%;存在200 ppm的SO2时的脱硝率为47.6%,下降了4.3%.
 |
| 图 6 SO2对氧活性物质注入协同SCR脱硝的影响(NOx 220 ppm,O2 6%,气体流速 1.5 L · min-1,SO2 200 ppm,电压 2.7 kV,空速 27200 h-1) Figure.6 Effect of SO2 on the NOx removal in the SCR coupled with ROS injection |
当含有SO2的烟气通过催化剂时,SO2一方面会在催化剂上与NH3形成竞争吸附;另一方面,吸附的SO2会与氨气反应形成亚硫酸铵,粘覆在金属氧化物催化剂的表面(Goo et al.,2007).因此,SO2会降低SCR脱硝性能.然而,本文研究表明,与常规标准SCR相比,采用氧活性物质注入协同快速SCR脱硝方法,SO2对脱硝性能的影响较小.这归因于O3氧化NO反应速率比氧化SO2的反应速率快了近8个数量级(Davis et al.,1974)(反应(12)),因此,O3对NO是选择性氧化(实验中也发现SO2浓度降低很小,低于5%),氧活性物质注入协同SCR脱硝过程并不会受SO3的影响;同时,氧活性物质注入协同SCR的脱硝速率远远快于标准SCR.因此,SO2对氧活性物质注入协同SCR脱硝过程的影响相对较小.
| SO2+O3→SO3+O2 | (12) |
图 7是烟气中存在水蒸汽(H2O)时,SCR脱硝率与烟气温度的关系.从图中可以看出,在温度150 ℃条件下,烟气中不含H2O时,氧活性物质注入协同SCR的脱硝效率是51.9%;而当模拟烟气中存在3%的H2O时,氧活性物质注入协同SCR的脱硝效率为45%,水蒸气的存在降低了SCR脱硝效率,但仍明显高于采用标准SCR时23.5%的脱硝率.
 |
| 图 7 H2O对氧活性物质注入协同SCR脱硝的影响(NOx 220 ppm,O2 6%,气体流速1.5 L · min-1,H2O 3%,电压2.7 kV,空速27200 h-1) Figure.7 Effect of H2O on the NOx removal in the SCR coupled with ROS injection |
在250 ℃以下,H2O能在催化剂表面通过水合作用与V2O5之间的形成稳定的氢键(Went et al.,1990).由于H2O是SCR的反应产物,也是烟气组分之一,当烟气中含有过量的H2O时,SCR脱硝反应的反应动力学常数必然会下降,脱硝反应受到抑制.Amiridis等(1996)通过Raman光谱实验,证明了H2O在催化剂表面能够通过与NH3形成竞争吸附抑制SCR脱硝效率.另外,H2O对快速SCR反应中间产物(NO-3、NO-2)的强烈吸附作用影响NO-3对V4+—OH的再氧化,也会影响NH3与NO-2之间直接脱硝反应.因此,烟气中H2O的存在降低了SCR脱硝效率.
4 结论(Conclusions)1)在温度低于300 ℃的低温区,采用氧活性物质注入方法将烟气中的NO部分氧化,NO/NO2浓度比为1时,可有效提高SCR的脱硝效率.
2)在150 ℃低温窗口下,烟气中存在200 ppm的SO2时,标准SCR脱硝效率下降了13.2%,采用氧活性物质注入协同SCR脱硝,脱硝率仅下降了4.3%.氧活性物质注入可以降低SO2对SCR脱硝性能的影响.
3)在烟气温度为150 ℃条件下,当模拟烟气中含3%的H2O时,氧活性物质注入协同SCR的脱硝效率为45%,明显高于采用标准SCR时23.5%的脱硝率.
| [1] | Amir idis M D,Wachs I E,Deo G,et al.1996. Reactivity of V2O5 catalysts for the selective catalytic reduction of NO by NH3: Influence of vanadia loading, H2O, and SO2 [J]. Journal of Catalysis, 161(1): 247-253 |
| [2] | 白希尧,沈丽,白敏的,等. 2000.强电离放电产生高浓度臭氧的基础理论与方法研究[J].物理, 29(10): 615-619 |
| [3] | Cheng Y S, Lambert C, Kim D H, et al. 2010. The different impacts of SO2 and SO3 on Cu/zeolite SCR catalysts [J]. Catalysis Today, 151(3/4): 266-270 |
| [4] | Davis D D, Prusazcyk J, Dwyer M, et al. 1974. Stop-flow time-of-flight mass spectrometry kinetics study. Reaction of ozone with nitrogen dioxide and sulfur dioxide [J]. The Journal of Physical Chemistry, 78(18): 1775-1779 |
| [5] | Goo J H, Irfan M F, Kim S D, et al. 2007.Effects of NO2 and SO2 on selective catalytic reduction of nitrogen oxides by ammonia[J]. Chemosphere, 67(4): 718-723 |
| [6] | Kim H H, Takashima K, Katsura S, et al. 2001. Low-temperature NOx reduction processes using combined systems of pulsed corona discharge and catalysts [J]. Journal of Physics (D: Applied Physics), 34(4): 604 |
| [7] | Koebel M, Elsener M, Kleemann M. 2000. Urea-SCR: a promising technique to reduce NOx emissions from automotive diesel engines [J]. Catalysis Today, 59(3/4): 335-345 |
| [8] | Lei Z G, Han B, Yang K, et al. 2013. Influence of H2O on the low-temperature NH3-SCR of NO over V2O5/AC catalyst: An experimental and modeling study [J]. Chemical Engineering Journal, 215-216: 651-657 |
| [9] | 林赫. 2002. 直流电晕放电诱导自由基簇射烟气脱硝试验和机理研究. 杭州: 浙江大学. 160 |
| [10] | Obradović B M, Sretenović G B, Kuraica M M. 2011. A dual-use of DBD plasma for simultaneous NOx and SO2 removal from coal-combustion flue gas [J]. Journal of Hazardous Materials, 185(2/3): 1280-1286 |
| [11] | 唐晓龙, 郝吉明, 徐文国, 等. 2005. 固定源低温选择性催化还原 NOx 技术研究进展 [J]. 环境科学学报, 25(10): 1297-1305 |
| [12] | Topsoe N Y, Dumesic J A, Topsoe H. 1995. Vanadia-titania catalysts for selective catalytic reduction of nitric-oxide by ammonia: I.I. studies of active sites and formulation of catalytic cycles [J]. Journal of Catalysis, 151(1): 241-252 |
| [13] | Tronconi E, Nova I, Ciardelli C, et al. 2007. Redox features in the catalytic mechanism of the "standard" and "fast" NH3-SCR of NOx over a V-based catalyst investigated by dynamic methods [J]. Journal of Catalysis, 245(1): 1-10 |
| [14] | Wang M Y, Zhu Tn L, Wang H. 2011. Oxidation and removal of NO from flue gas by DC corona discharge combined with alkaline absorption [J]. IEEE Transactions on Plasma Science, 39(2): 704-710 |
| [15] | 王新新. 2009. 介质阻挡放电及其应用[J]. 高电压技术, 35(1): 1-11 |
| [16] | Went G T, Oyama S T, Bell A T. 1990. Laser Raman spectroscopy of supported vanadium oxide catalysts [J]. The Journal of Physical Chemistry, 94(10): 4240-4246 |
| [17] | 谢维杰, 李龙海, 周保学, 等. 2008. 氧气常压介质阻挡放电的发射光谱及能量传递机理[J]. 物理化学学报, 24(5): 827-832 |
| [18] | 张烨, 缪明烽. 2011. SCR脱硝催化剂失活机理研究综述[J]. 电力科技与环保, 27(6): 6-9 |