 2014, Vol. 34
2014, Vol. 34


 , 杜荣斌1, 刘涛1
, 杜荣斌1, 刘涛1 2. 山西大学 环境与资源学院, 太原 030006
2. College of Environmental Science and Resources, Shanxi University, Taiyuan 030006
燃煤烟气排放的Hg会对环境和人体造成严重、持久的危害,目前已引起人们的广泛关注(Golding et al., 2007;Fitzgerald et al., 1998).尽管汞在煤中的含量很低,但由于煤的用量巨大,燃煤Hg排放成为环境汞污染的重要来源,约占全球每年人为汞排放量的30%(Hsi et al., 1998).随着燃煤Hg污染问题的逐渐加重及人们环保意识的日益增强,世界各国政府相继颁布了法律法规来限制燃煤电厂的汞排放.美国国家环境保护局(EPA)于2005年制定了清洁空气Hg排放法规来限制和减少燃煤电厂Hg的排放(U.S.EPA,2005).
我国是能源消耗大国,煤炭在一次能源结构中所占比例很大,且在今后相当长的一个时期内难以改变,预计到2015年占63%,2050年煤炭的利用仍将占50%以上(谢克昌,2009).我国煤炭利用以直接燃烧(发电和供热)为主,因此,燃煤Hg污染问题更加突出,不容忽视.我国在2012年1月1日发布了新修订的《火电厂大气污染物排放标准》(GB13223—2011),对火电厂汞的排放做出了明确规定,将烟气中汞的排放值限定在0.03 mg·m-3.
燃煤烟气中Hg的形态主要有3种:气态Hg0(Hg0g)、气态Hg2+(Hg2+g)和吸附于飞灰上的Hgp(Galbreath et al., 2000).其中,Hg2+g易溶于水,可以较容易地通过湿式除尘或湿法脱硫装置脱除,Hgp能随飞灰一起被除尘装置脱除.Hg0g占燃煤烟气中气态Hg的20%~50%(Senior et al., 2000),由于其挥发性高和难溶于水的特性,难以被现有的污染物控制装置脱除,因而成为燃煤烟气Hg污染控制研究的重点和难点,但目前尚未形成经济有效的控制技术.
目前研发的燃煤Hg0排放控制方式主要是在烟道中喷入活性炭粉末吸附气态Hg0,将Hg0g转变为Hgp后再经除尘装置脱除.但此方法的工业应用仍存在一些问题需要解决,如活性炭对Hg0g的吸附主要是物理吸附,受烟气温度及烟气成分的影响很大.特别是活性炭喷射需在除尘前进行,烟气的高温不利于活性炭对Hg0g的吸附(Miller et al., 2000;Feeley et al., 2005),使得活性炭对Hg0g的吸附能力较差,导致活性炭的利用率低、消耗量大,运行成本很高(Jones et al., 2007).因此,经济有效的燃煤Hg0g污染控制技术亟待开发利用.
研究表明,Fe2O3对Hg0g具有一定的氧化活性,可将Hg0g氧化为较易脱除的Hg2+g,这主要是由于铁具有多价性,电子容易迁移,因此,具有较高的催化氧化活性(Kamata et al., 2009;Wu et al., 2008).粉煤灰是燃煤烟气中收捕下来的细灰,也是燃煤电厂排出的主要固体废物,研究发现,其对烟气中的Hg0g具有一定的吸附能力,特别是粉煤灰中所含的Fe2O3对其吸附Hg0g具有促进作用,且受到粉煤灰粒径及其主要化学成分等因素影响(Donatello et al., 2012;Li et al., 2012;Kostova et al., 2011;Abad-Valle et al., 2011;Dunham et al., 2003).因此,本研究通过负载Fe2O3到粉煤灰制备Fe2O3/FA催化剂用于脱除气态Hg0,研究Fe2O3负载量、温度、空速、Hg0浓度和烟气成分等对Fe2O3/FA脱除Hg0的影响,并对Fe2O3/FA上吸附Hg的形态进行分析表征.
2 材料与方法(Materials and methods) 2.1 催化剂制备粉煤灰来源于中石化安庆分公司热电厂,主要化学成分为48.29% SiO2、21.64% Al2O3、3.05% Fe2O3、20.33% CaO、4.37% C,实验所用粒径为250~350 μm.将粉煤灰等体积浸渍在一定浓度的Fe(NO3)3溶液中,在室温静置2 h,然后依次在50 ℃干燥5 h,110 ℃干燥5 h,最后在一定温度Ar气氛中煅烧5 h,制备了Fe2O3负载量为1%~10%的Fe2O3/FA催化剂,并根据Fe2O3负载量标记为Fex/FA,如Fe2O3负载量为10%的Fe2O3/FA标记为Fe10/FA.
2.2 Hg0的脱除实验Hg0的脱除实验在固定床石英管反应器中进行(图 1),催化剂装填量为0.2 g,反应温度为120~200 ℃.实验气氛为N2+6.3% O2和含有一定浓度烟气成分的N2,Hg0浓度为420 μg·m-3,气体总流量为105 mL·min-1,空速约为15000 h-1.Hg0蒸气由汞渗透管(美国VICI 公司)产生,通过改变水浴温度及载气流量,可以产生不同浓度的Hg0蒸气.催化剂在反应器中程序升温至设定温度后,开始通入含Hg0的N2+O2或含有一定浓度烟气成分的N2进行恒温吸附,吸附时间为4 h.反应器出口气体通过一个常温活性炭床层后排空.
 |
| 图 1 Hg0脱除实验装置(1.转子流量计;2.质量流量计;3.水浴;4.气体混合室;5.加热带;6.汞渗透管;7.反应器;8.电炉;9.石英棉;10.催化剂;11.温度控制仪;12.活性炭) Fig. 1 Schematic diagram of Hg0 removal apparatus |
催化剂的Hg吸附量为脱除Hg0前、后催化剂上Hg含量的差值,根据中国国家标准GB/T 16659—1996,采用H2SO4+HNO3对样品进行消解后,经原子荧光光谱仪(AFS)测得.为了讨论方便,本文引入脱除效率E的概念,定义为一定时间内催化剂上吸附的Hg0量与进入反应器的Hg0量的百分比,即:
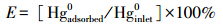
采用逐级化学提取实验测定吸附Hg0后FA和Fe2O3/FA样品上Hg的形态(Bloom et al., 2003).将吸附Hg0后样品依次经过5个溶液进行化学提取:二次去离子水可提取水溶性的汞化合物;0.1 mol·L-1 CH3COOH+0.01 mol·L-1 HCl可提取可溶于“胃酸”溶液的汞化合物;1 mol·L-1 KOH可提取有机汞;12 mol·L-1 HNO3可提取元素汞;王水可提取汞的硫化物和硒化物.液固比为100/1,样品在室温下不断搅拌提取18~22 h.样品依次经过一种提取溶液提取2次,提取液中的Hg含量利用原子荧光光谱仪测定.本文中的实验数据均为3次测量的平均值,相对标准偏差小于5%.
3 结果与讨论(Results and discussion) 3.1 Fe2O3/FA对Hg0的脱除图 2为N2+O2气氛中,不同Fe2O3负载量的Fe2O3/FA在温度为150 ℃、Hg0浓度为420 μg·m-3条件下对Hg0脱除4 h的结果.可以看出,载体FA对Hg0的脱除能力较低,4 h的脱除效率仅为17%,这缘于FA对Hg0的吸附主要是物理吸附的结果(Abad-Valle et al., 2011;Dunham et al., 2003).Fe2O3/FA对Hg0的脱除能力明显高于FA,这显然是与其上负载的Fe2O3有关,可能主要缘于Fe2O3对Hg0的催化氧化作用(Kamata et al., 2009;Wu et al., 2008),这与文献中报道的Fe2O3对Hg0具有氧化作用的结果一致.此外,随着Fe2O3负载量的增加,Fe2O3/FA对Hg0的脱除能力增强.Fe2O3的负载量由1%增加到10%,Fe2O3/FA对Hg0的脱除效率由26%增高到45%.因此,以下实验以Fe2O3负载量为10%的Fe2O3/FA为例,考察了Fe2O3/FA在不同的反应温度、空速、Hg0浓度和烟气成分下对Hg0的脱除.
 |
| 图 2 Fe2O3/FA与FA对Hg0的脱除能力对比 Fig. 2 Comparison of Hg0 removal capability by Fe2O3/FA and FA |
图 3为Fe10/FA在不同反应温度(120、150、180和200 ℃)条件下对Hg0脱除4 h的结果.可以看出,Fe2O3/FA对Hg0的脱除能力随温度的升高而增强.温度从120 ℃升高到150 ℃,Fe2O3/FA对Hg0的脱除效率增幅较大,由31%升高到45%.继续升高温度至180 ℃和200 ℃,Hg0的脱除效率分别升高至48%和49%,增幅较小.因催化剂制备过程的煅烧温度(300 ℃)远高于实验温度(120~200 ℃),粉煤灰的孔隙结构在Fe2O3/FA对Hg0的脱除过程中不会发生明显变化,因此,Fe2O3/FA对Hg0的脱除效率随温度变化的趋势可能缘于Fe2O3/FA对Hg0的脱除是Fe2O3的氧化作用和FA的吸附作用的共同结果.随温度升高,Fe2O3的氧化活性会增强,而FA的吸附作用会减弱(包括FA上吸附Hg的脱附),两者在不同温度下发挥的作用程度不同而构成了Fe2O3/FA对Hg0的脱除结果.图 3中Hg0的脱除效率随温度的变化趋势表明,在排烟温度(120~200 ℃)范围内,Fe2O3的氧化作用在Fe2O3/FA对Hg0的脱除过程中起到了主要作用,这与Wang等(2012)的研究结果是一致的.
 |
| 图 3 温度对Fe2O3/FA脱除Hg0的影响 Fig. 3 Effect of temperature on Hg0 removal by Fe2O3/FA |
图 4为Fe2O3/FA在温度为150 ℃、空速分别为5000、8000、12000和15000 h-1条件下对Hg0脱除4 h的结果.可以看出,随着空速降低,Fe2O3/FA对Hg0的脱除效率提高.空速由15000 h-1降低至5000 h-1,Fe2O3/FA对Hg0的脱除效率由45%升高至54%.这主要是由于空速降低,增加了Fe2O3/FA与Hg0的接触时间,提高了Fe2O3/FA催化剂表面上Fe2O3的利用率,进而提高了Fe2O3/FA对Hg0的脱除效率,这与文献中报道的空速对Fe2O3/AC脱除SO2和CuO/AC脱除Hg0的影响一致(马建蓉等,2001;Wang et al., 2013).图 4中的结果表明,Fe2O3/FA在常规的工业烟气净化运行空速条件下,具有良好的脱除Hg0的效果.
 |
| 图 4 空速对Fe2O3/FA脱除Hg0的影响 Fig. 4 Effect of space velocity on Hg0 removal by Fe2O3/FA |
为了进一步评价Fe2O3/FA在实际燃煤烟气所含的Hg0浓度范围内(1~35 μg·m-3)(Serre et al., 2000)对Hg0的脱除能力,进行了Fe2O3/FA在温度为150 ℃、不同Hg0浓度(21、195、250和420 μg·m-3)条件下脱除Hg0的实验,结果示于图 5.可以看出,较低的Hg0浓度条件下,Fe2O3/FA获得了更高的脱除Hg0的效率.Hg0浓度由420 μg·m-3降低到 21 μg·m-3,Fe2O3/FA对Hg0的脱除效率由45%升高到62%.这表明Fe2O3/FA在较宽的Hg0浓度范围内具有较高的脱除Hg0的能力,在实际燃煤烟气所含的Hg0浓度范围内具有良好的脱除Hg0的效果.
 |
| 图 5 Hg0浓度对Fe2O3/FA脱除Hg0的影响 Fig. 5 Effect of Hg0 concentration on Hg0 removal by Fe2O3/FA |
燃煤烟气中的重要成分O2、SO2、HCl、H2O可能会对Fe2O3/FA脱除Hg0产生影响.因此,图 6对比了N2、N2+6.3% O2、N2+1500 ppm SO2、N2+50 ppm HCl、N2+5% H2O气氛中,Fe2O3/FA在温度为150 ℃条件下对Hg0脱除4 h的结果.可以看出,HCl和O2对Fe2O3/FA脱除Hg0具有促进作用,而SO2和H2O抑制了Fe2O3/FA对Hg0的脱除.HCl对Fe2O3/FA脱除Hg0的促进作用可能缘于其可吸附在Fe2O3活性位上,进而与气相中或吸附在Fe2O3/FA上的Hg0反应,从而增强了Hg0的氧化(Wu et al., 2008).O2的促进作用与文献中报道的其对金属氧化物氧化Hg0的促进作用是一致的,主要是通过向氧化Hg0后失去氧化活性的金属氧化物提供氧而使其恢复氧化活性(Wang et al., 2010;Presto et al., 2006). SO2的抑制作用主要是由于其可吸附到Fe2O3活性位上并与之反应生成Fe2(SO4)3,消耗了可吸附氧化Hg0的Fe2O3活性位(马建蓉等,2001;Ma et al., 2003).H2O的抑制作用主要是由于其可吸附到Fe2O3/FA上,与Hg0在Fe2O3活性位上存在竞争吸附,影响了Hg0的吸附并进而影响了Hg0的氧化(Ma et al., 2003).
 |
| 图 6 烟气成分对Fe2O3/FA脱除Hg0的影响 Fig. 6 Effect of flue gas components on Hg0 removal by Fe2O3/FA |
为了判明Fe2O3对Hg0的氧化作用,确定Fe2O3/FA上吸附Hg的形态及各形态Hg的含量,对脱除Hg0后的FA(FA-Hg)和Fe2O3/FA(Fe2O3/FA-Hg)样品进行了逐级化学提取实验,结果示于图 7.可以看出,FA-Hg上的Hg主要是在HNO3中被提取出来,大约占到FA-Hg上提取出的总Hg量的85%,而Fe2O3/FA-Hg上的Hg主要是在CH3COOH+HCl中被提取出来,约占到Fe2O3/FA-Hg上提取出 的总Hg量的63%,同时二次去离子水也提取出了约8%的Hg.根据文献(Bloom et al., 2003),HNO3中提取出的Hg主要是Hg0,而CH3COOH+HCl与二次去离子水中提取出的Hg主要是Hg2+的化合物,表明FA上吸附的Hg主要是以元素态Hg0的形式存在而Fe2O3/FA-Hg上吸附的Hg主要是以Hg2+的化合物形式存在,约为71%.因本实验所采用的气氛中只有N2+O2,因此,Fe2O3/FA上吸附的Hg2+化合物只有可能是HgO.此外,对于Fe2O3/FA-Hg样品,在HNO3中也提取出部分Hg(约24%),说明Fe2O3/FA上吸附的Hg也包含部分Hg0.由此可以判明,Fe2O3在Fe2O3/FA脱除Hg0过程中起了关键作用,Fe2O3将Hg0催化氧化为Hg2+的化合物HgO并吸附在FA上,从而显著提高了Fe2O3/FA对Hg0的脱除能力.
 |
| 图 7 脱除Hg0后FA和Fe2O3/FA样品的逐级化学提取 Fig. 7 Sequential chemical extraction of Hg0-captured FA and Fe2O3/FA samples |
1)负载Fe2O3到粉煤灰显著提高了其对Hg0的脱除能力,主要缘于Fe2O3的氧化活性,将Hg0氧化为HgO并吸附在Fe2O3/FA上.
2)在排烟温度及燃煤烟气所含的Hg0浓度范围内,Fe2O3/FA具有良好的脱除Hg0的能力,且随Fe2O3负载量的增加及温度的升高而增强.在常规的工业烟气净化运行空速条件下,Fe2O3/FA展现了良好的脱除Hg0的能力.
3)HCl和O2对Fe2O3/FA脱除Hg0具有促进作用,而SO2和H2O抑制了Fe2O3/FA对Hg0的脱除.
| [1] | Abad -Valle P,Lopez-Anton M,Diaz-Somoano M,et al.2011.The role of unburned carbon concentrates from fly ashes in the oxidation and retention of mercury[J].Chemical Engineering Journal,174(1): 86-92 |
| [2] | Bloom N S,Preus E,Katon J,et al.2003.Selective extractions to assess the biogeochemically relevant fractionation of inorganic mercury in sediments and soils[J].Analytica Chimica Acta,479(2): 233-248 |
| [3] | Donatello S,Fernández-Jiménez A,Palomo A.2012.An assessment of mercury immobilisation in alkali activated fly ash (AAFA) cements[J].Journal of Hazardous Materials,213-214: 207-215 |
| [4] | Dunham G E,DeWall R A,Senior C L.2003.Fixed-bed studies of the interactions between mercury and coal combustion fly ash[J].Fuel Processing Technology,82(2/3): 197-213 |
| [5] | Feeley T,Brickett L.2005.Field testing of mercury control technologies for coal-fired power plants[R].DOE Mercury R&D Program Review,U.S.DOE |
| [6] | Fitzgerald W F,Engstrom D R,Mason R P,et al.1998.The case for atmospheric mercury contamination in remote areas[J].Environmental Science & Technology,32(1): 1-7 |
| [7] | Galbreath K C,Zygarlicke C J.2000.Mercury transformations in coal combustion flue gas[J].Fuel Processing Technology,65-66: 289-310 |
| [8] | Golding G R,Kelly C A,Sparling R,et al.2007.Evaluation of mercury toxicity as a predictor of mercury bioavailability[J].Environmental Science & Technology,41(16): 5685-5692 |
| [9] | Hsi H C,Chen S G,Rostam-Abadi M,et al.1998.Preparation and evaluation of coal-derived activated carbons for removal of mercury vapor from simulated coal combustion flue gases[J].Energy & Fuels,12(6): 1061-1070 |
| [10] | Jones A P,Hoffmann J W,Smith D N,et al.2007.DOE/NETL’s phase Ⅱ mercury control technology field testing program: Preliminary economic analysis of activated carbon injection[J].Environmental Science & Technology,41(4): 1365-1371 |
| [11] | Kamata H,Ueno S,Sato N,et al.2009.Mercury oxidation by hydrochloric acid over TiO2 supported metal oxide catalysts in coal combustion flue gas[J].Fuel Processing Technology,90(7/8): 947-951 |
| [12] | Kostova I,Hower J,Mastalerz M,et al.2011.Mercury capture by selected Bulgarian fly ashes: Influence of coal rank and fly ash carbon pore structure on capture efficiency[J].Applied Geochemistry,26(1): 18-27 |
| [13] | Li J R,Maroto-Valer M.2012.Computational and experimental studies of mercury adsorption on unburned carbon present in fly ash[J].Carbon,50(5): 1913-1924 |
| [14] | 马建蓉,刘守军,刘振宇,等.2001.新型低温Fe/AC脱硫剂的研究[J].环境科学,22(6): 29-33 |
| [15] | Ma J R,Liu Z Y,Liu S J,et al.2003.A regenerable Fe/AC desulfurizer for SO2 adsorption at low temperatures[J].Applied Catalysis B: Environmental,45(4): 301-309 |
| [16] | Miller S J,Dunham G E,Olson E S,et al.2000.Flue gas effects on a carbon-based mercury sorbent[J].Fuel Processing Technology,65-66: 343-364 |
| [17] | Presto A A,Granite E J.2006.Survey of catalysts for oxidation of mercury in flue gas[J].Environmental Science & Technology,40(18): 5601-5609 |
| [18] | Senior C L,Sarofim A F,Zeng T F,et al.2000.Gas-phase transformations of mercury in coal-fired power plants[J].Fuel Processing Technology,63(2/3): 197-213 |
| [19] | Serre S D,Silcox G D.2000.Adsorption of elemental mercury on the residual carbon in coal fly ash[J].Industrial & Engineering Chemistry Research,39(6): 1723-1730 |
| [20] | U.S.EPA.2005.Clean Air Mercury Rule,40 CFR Parts 60,63,72 and 75[P].U.S.EPA,Washington,DC |
| [21] | Wang J W,Kong X J,Du R B,et al.2013.Removal of vapor-phase elemental mercury over a CuO/AC catalyst[J].Advanced Materials Research,610-613: 64-67 |
| [22] | Wang J W,Liu R Q,Kong X J.2012.Gas-phase Hg0 removal by a Fe2O3/AC catalyst[J].Advanced Materials Research,518-523: 210-213 |
| [23] | Wang J W,Yang J L,Liu Z Y.2010.Gas-phase elemental mercury capture by a V2O5/AC catalyst[J].Fuel Processing Technology,91(6): 676-680 |
| [24] | Wu S J,Ozaki M,Uddin M A,et al.2008.Development of iron-based sorbents for Hg0 removal from coal derived fuel gas: Effect of hydrogen chloride[J].Fuel,87(4/5): 467-474 |
| [25] | 谢克昌.2009.煤炭的低碳化转化和利用[J].山西能源与节能,1: 1-3 |