 2014, Vol. 34
2014, Vol. 34



湿法烟气脱硫(WFGD)技术是目前世界上最为有效可靠的脱硫工艺之一(Brogren et al., 1997),其中,石灰石/石膏湿法脱硫技术应用最为广泛,截止到2011年底,占我国已投产脱硫机组份额的92%以上.2011年新颁布的《火电厂大气污染物排放标准》(GB132323—2011)对我国火电厂SO2排放提出了更高要求,SO2排放限值降为100 mg · Nm-3,重点地区排放限值降为50 mg · Nm-3.国内现行脱硫工艺面临新的挑战,燃煤锅炉原有脱硫设备大部分将面临升级改造(中国环境保护产业协会脱硫脱硝委员会,2013).因此,进一步提升脱硫效率,开发优化石灰石/石膏湿法烟气深度脱硫技术势在必行.
了解脱硫浆液内组分传质、溶解、反应等机理及深入优化脱硫温度等运行参数是进一步开发深度脱硫技术的关键.近年来,国内外学者针对脱硫过程中SO2、石灰石等组分吸收溶解机制进行了一系列深入研究(Brogren et al., 1997;Kiil et al., 1998;Wang et al., 2008;Souza et al., 2010),研究方法主要集中于实验研究、数值计算、工业试验等.由于目前测量手段或数值计算方法的局限性,研究对象的时空尺度主要侧重于微秒及微米以上,针对更小时空尺度的研究相对较少.
分子动力学(Molecular Dynamics,MD)是一种新兴的理论化学计算方法,能够从纳米尺度及纳秒尺度甚至更小的时空尺度深入研究气液/溶液中的分子微观结构、传质扩散机制、反应机理等(Garrett et al., 2006).针对N2、O2、CO2、H2O、NH3等小分子气体在水中的溶解机理人们已经运用分子动力学进行了系统的研究(Somasundaram et al., 1999;cha et al., 2004;Paul et al., 2005;Dang et al., 2004).但针对SO2溶解机制的研究相对较晚,部分先驱者初步研究了SO2在纯水表面的反射、吸收行为(Matsumoto,1996),以及水溶液内SO2周围H2O朝向、氢键等微观结构、SO2溶解副产物对SO2溶解的耦合影响(Tarbuck et al., 2006)等.Shamay等(2011)进一步研究了SO2在气液界面的迁移途径及分布梯度,Moin等(2011)结合QM-MD方法研究了SO2在水溶液中的水化结构及SO2-H2O作用位等,研究发现,SO2表面形成了一层稳定的水化层.
前人主要以水溶液作为液相进行研究,针对含CaCO3等吸收剂及其离子的浆液体系的研究还相对缺乏.从分子层面研究SO2等组分在浆液中的传质机制及溶解吸收行为能够进一步为钙基湿法脱硫反应机理的深入研究及脱硫工艺优化提供有力的理论依据,具有重要的现实意义.基于此,本文将利用分子动力学计算研究浆液内组分扩散系数及SO2在浆液中的溶解机制,并进一步分析脱硫运行温度对组分扩散系数及SO2溶解吸收的影响作用.本文研究结果将为深入了解浆液内SO2溶解微观结构及筛选脱硫运行温度提供有效理论依据.
2 计算模型及方法(Computational models and simulation method)本文研究的SO2-浆液体系主要针对液相内,不考虑气相及未溶解的固相的影响,故溶液元胞模型采用正方体的模拟盒子.由于钙基湿法脱硫吸收剂中的主要作用成分为CaCO3,又考虑实际浆液环境中HCO-3、HSO-3等两性离子的复杂反应机制及力场适用性,为简化计算,浆液模型设计中只考虑Ca2+和CO2-3两种离子组分.石灰石-石膏湿法脱硫浆液中溶解盐的质量分数在1.2%左右,根据浓度换算结果,在模拟体系中加入864个H2O分子、2个Ca2+和2个CO2-3来构建所需浆液模型.脱硫工艺稳定运行过程中,浆液单位时间的SO2吸收量可以通过计算排出的固渣中的含硫物含量得到,以脱硫浆液内含固量约20%(王乃华等,2005)且其中90%为CaSO4 · H2O的条件计算,折合SO2吸收量为约1.5 mol · L-1.进一步考虑本文SO2溶解特性的研究初衷,在“浆液”盒子中随机插入16个SO2分子,构成SO2吸收浓度近似为1 mol · L-1的溶液体系.为了消除边界效应,盒子的3个方向都采用了周期性条件.初始盒子的尺寸为3.032 nm×3.032 nm×3.032 nm.本文中,H2O分子的势能函数采用了著名的SPC模型(Berendsen et al., 1981).Ca2+、CO2-3、SO2的分子/离子模型由Material Studio软件(Accelrys Inc.)构建优化,各原子的力场参数及电荷分布由COMPASS力场赋予,该力场已分别被证实适用于这3种粒子的溶液/液相特性计算(Yuan et al., 2013;Aguiar et al., 2013;Yang et al.,2000).
体系构建完成后,先对系统用Smart Minimizer法进行能量最小化处理;随后在等温等压系综(NPT)下将体系进行300~500 K、100 ps的退火弛豫以消除盒子内的局部应力并找到全局能量最低构象,退火弛豫循环5次,升温及降温过程中每次温差变化为10 K.然后对最优构象进行150 ps的NPT平衡弛豫,当系统势能波动在5%以内,计算体系达到稳定.最后对稳定构象进行600 ps的正则系综(NVT)模拟计算,每0.5 ps收集一次各原子的动力学计算全轨迹.为了消除计算运行的初始影响,取后500 ps的数据进行性质分析.NPT系综下运行的平衡压力为1 atm(1.01×105 Pa),NPT系综和NVT系综下的运行温度均为计算目标温度.
分子动力学运行计算中,选用的力场为COMPASS通用力场,NPT系综弛豫及NVT系综模拟中温度控制采用Andersen热浴法(Andersen,1980),压力控制采用Bererdsen恒压法(Berendsen et al., 1984).计算非键作用中,范德华作用(van der Waals)采用Atom based方法(Karasawa et al., 1992),截断半径为1.25 nm,静电作用采用Ewald方法(Allen et al., 1989).各分子的初始运动速度采用Maxwell-Boltzman分布随机取样,采用Velocity-verlet积分方法进行求解,时间步长为1fs.所有计算模拟过程均在Material Studio软件(Accelrys Inc.)上进行.
石灰石-石膏湿法脱硫工艺的运行温度在50 ℃左右,因此,本文设计的模拟温度为293.15~343.15 K,每间隔10 K计算1次.
3 结果与分析(Results and discussion)图 1给出了293.15 K运行温度下的模型例图,其中,线性分子模型为H2O分子,黄红球棍模型为SO2分子,灰红球棍模型为CO2-3,绿色小球为Ca2+.经过不同温度的等温等压系综(NPT)平衡弛豫后,模拟体系密度达到稳定.在计算温度范围内随温度的升高293.15~343.15 K,体系密度缓慢下降(1.005~0.952 g · cm-2),与实际情况下水密度接近(CaCO3溶质的影响可以忽略),表明计算可靠(图 2).
 |
| 图 1 SO2吸收量为1 mol · L-1的溶液模型(以293.15 K为例) Fig. 1 A Snapshot of the cell structure of 1 mol · L-1 aqueous solutions of SO2(model of 293.15 K as a example) |
 |
| 图 2 模型密度的模拟结果与实际密度的对比 Fig. 2 Plot of simulated cell density vs. experimental water density |
溶液中组分的扩散系数是表征体系内各物质传质输运特性的重要参数.湿法脱硫反应中,气液传质阻力主要来源于液相(钟毅,2008),溶液组分扩散系数是影响整个脱硫反应工艺传质能力、脱硫反应速率及脱硫效率等的关键因素.分子动力学中计算粒子的扩散系数可以通过求解利用该粒子的均方位移(MSD,Mean Square Displacement)的Eintein关系式获得,即:
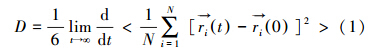
表 1中详细列出了293.15~343.15 K运行温度下H2O、SO2、Ca2+、CO2-3 4种粒子的扩散系数计算结果.溶液中SO2、Ca2+、CO2-3等粒子扩散系数一般由实验测得(丁红蕾,2010;Himmelblau,1964;Ebanks et al., 2010),通过MD计算的文献较少.计算发现,H2O分子扩散系数从293.15 K时的3.074×10-9m2 · s-1逐渐上升到343.15 K时的6.079×10-9 m2 · s-1.Mark等(2001)对SPC、TIP3P、SPC/E等多个H2O分子模型及修正模型在298 K下的扩散系数进行了计算,结果分别为4.22×10-9 m2 · s-1(SPC)、2.75×10-9 m2 · s-1(SPC/E)、5.67×10-9 m2 · s-1(TIP3P),本文计算结果与该文献符合较好.溶液中SO2扩散系数的计算结果与文献中参考值(20~40 ℃,1.62×10-9 ~2.59×10-9 m2 · s-1,Himmelblau,1964;25 ℃,1.83×10-9 m2 · s-1,丁红蕾,2010)符合较好.而Ca2+和CO2-3的计算结果则与文献(Chen et al., 2013;Bruneval et al., 2007)的研究结果较为接近.
| 表1 293.15~343.15 K温度下溶液组分的扩散系数计算结果 Table 1 Computed diffusion coefficient of components in aqueous 293.15~343.15 K |
表 1中4种粒子的扩散系数均随温度的升高而升高,这是由于温度升高,粒子内能增加,动能增大,扩散能力增强.进一步而言,粒子之间碰撞几率加大,有效碰撞机会增多,从而加速脱硫反应.H2O分子在溶液中是以氢键结合的分子团簇形式存在,温度升高使得H2O分子在团簇内平衡位置振动增强,氢键结构被破坏,自由H2O分子增多.这也会增大依赖于H2O分子扩散的粒子的扩散能力,如H+等(Walbran et al., 2001),进一步加大了脱硫反应速率.
溶液中组分扩散系数与温度的关系应符合扩散动力学的Arrhenius定律,即lnD=lnDo-ΔE/(RT),其中,Do为指前因子(m · s-1),ΔE为扩散系数的表观活化能(kg · mol-1),R为气体常数,取值为8.314 J · mol-1,T为温度(K).图 3中给出了H2O、SO2、Ca2+、CO2-3 4种粒子的扩散系数的对数lnD与温度的倒数1/T的关系曲线.可以看到,4种粒子的lnD-l/T曲线线性较好.根据公式lnD=lnDo-ΔE/(RT)计算得到了4种粒子的扩散活化能,其中,H2O分子的扩散活化能为11.27 kJ · mol-1,SO2的扩散活化能为11.47 kJ · mol-1,Ca2+和CO32-的扩散活化能分别为16.74 kJ · mol-1和21.32 kJ · mol-1.
 |
| 图 3 溶液组分扩散系数与温度的lnD-1/T曲线(293.15~343.15K) Fig. 3 Plot of lnD vs. 1/T of the diffusion coefficient of components in aqueous(293.15~343.15K) |
溶液内溶质分子或离子与溶剂分子产生相互结合,称为溶剂化现象,水溶液中这种现象又称为水化现象.溶质水化对溶质溶解、溶质扩散等都有重要影响,研究湿法脱硫中SO2在浆液中水化、溶解的微观机制十分必要.分子动力学中分析溶液内粒子间微观结构的直接有效手段之一是分析径向分布函数(Radical Distribution Function,RDF)(Moin et al., 2011;Kerisit et al., 2005;Bruneval et al., 2007).通过径向分布函数曲线可以获得溶液体系内粒子间相互作用、粒子间有序性等,还可以进一步计算得到粒子间的配位能力,具体计算方法如下(Allen et al., 1989):
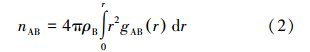
 |
| 图 4 不同温度下(293.15~343.15 K)S(SO2)-Ow径向分布函数及配位数 Fig. 4 Radical distribution functions of S(SO2)-Ow and running integration numbers for different temperatures(293.15~343.15 K) |
图 4给出了不同温度下293.15~343.15 K模拟体系内SO2中S原子与周围的H2O中O原子的径向分布函数(S(SO2)-Ow RDF),并由该径向分布函数进一步计算得到了SO2在不同温度的浆液内的H2O配位数变化情况,具体见图 4右上角附图.S(SO2)-Ow RDF中只有一个明显单峰,第一峰位于约0.4225 nm处,第一峰谷位于0.5625 nm处,表明浆液内SO2与H2O之间只形成一个相对明显的水化层,这与Moin等(2011)的研究结果一致.不同温度下293.15~343.15 K第一峰与第一峰谷的位置基本没有变化,即温度对SO2与H2O结合的局部结构没有太大影响.峰值大小可以表征SO2与周围H2O之间作用的强弱程度,313.15 K与323.15 K下计算峰值最高,分别达到1.372与1.347,而其他4种温度下则峰值相对较低.进一步分析发现,SO2周围的H2O配位数在计算温度区间内形成明显单峰分布,313.15 K与323.15 K时H2O配位数最大,分别达到20.30与19.89,即313.15~323.15 K之间SO2与H2O作用最强,最有利于SO2溶解.
为了进一步研究浆液中Ca2+对SO2的作用机制,图 5中给出了Ca2+与S(SO2)的径向分布函数.RDF曲线中Ca2+与SO2并没有形成稳定的峰型,只存在一个模糊单峰,表明模拟浆液中Ca2+与SO2并没有形成稳定结构,Ca2+与SO2之间的相互作用主要为范德华作用.313.15 K模拟温度下峰值最高,即323.15 K工况下Ca2+对SO2作用最强,最利于吸收反应.这可能与本文之前分析发现的313.15~323.15 K温度下SO2最易溶解有关.根据湿法脱硫工艺反应原理,SO2溶解在浆液中并与H2O反应形成H2SO3,H2SO3电离形成HSO-3、SO2-3等离子,Ca2+与这些离子相互结合然后进行化学反应.SO2溶解的难易程度决定了其与Ca2+的作用强弱.
 |
| 图 5 不同温度下(293.15~343.15 K)Ca2+-S(SO2)径向分布函数 Fig. 5 Radical distribution functions of Ca2+-S(SO2)for different temperatures(293.15~343.15 K) |
通过引入相互作用能(Interaction Energy,IE)概念(Hanus et al., 2006;廖瑞金等,2012)进一步定量分析了温度对浆液中SO2扩散、溶解能力的影响机制.依照相互作用能概念,本文定义如下计算式:
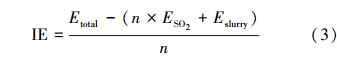
 |
| 图 6 不同温度下(293.15~343.15 K)SO2-浆液的相互作用能 Fig. 6 SO2-Slurry interaction energies for different temperatures(293.15~343.15 K) |
本文利用分子动力学方法研究了钙基湿法脱硫中浆液组分的扩散行为及SO2在浆液中的溶解机制,并通过计算不同温度模型293.15~343.15 K,分析获得了温度对脱硫浆液组分扩散能力及SO2溶解能力的影响规律.所得结论如下:
1)通过不同温度293.15~343.15 K下浆液模型的模拟计算,得到了浆液中H2O、SO2、Ca2+、CO2-3 4种粒子在计算温度区间内的扩散系数,分别为3.074×10-9~6.079×10-9 m2 · s-1(H2O)、1.649×10-9~3.198×10-9 m2 · s-1(SO2)、 0.3157×10-9~0.8863×10-9 m2 · s-1(Ca2+)及0.2516×10-9~0.9674×10-9 m2 · s-1(CO2-3).4种粒子的扩散系数均随温度升高而增大.进一步通过Arrhenius公式计算得到了这4种粒子的扩散活化能,分别为11.27、11.47、16.74及21.32 kJ · mol-1.
2)浆液内SO2与H2O之间只形成一个相对明显的水化层;SO2周围的H2O配位数在293.15~343.15 K计算温度下内呈单峰分布;313.15~323.15 K时SO2水化作用最强,最利于SO2溶解.
3)进一步的SO2-浆液相互作用能分析表明,293.15~343.15 K温度内随温度升高,浆液与SO2结合能力增大;而323.15 K左右浆液对SO2捕集能力最强.
| [1] | Aguiar J E,Bezerra B T C,De Melo Braga B,et al.2013.Adsorption of anionic and cationic dyes from aqueous solution on non-Calcined Mg-Al layered double hydroxide: Experimental and theoretical study[J].Separation Science and Technology,48(15): 2307-2316 |
| [2] | Allen M P,Tildesley D J.1989.Computer Simulation of Liquids[M].Britain: Oxford University Press |
| [3] | Andersen H C.1980.Molecular dynamics simulations at constant pressure and/or temperature[J].The Journal of Chemical Physics,72(4): 2384-2393 |
| [4] | Berendsen H J C,Postma J P M,Van Gunsteren W F,et al.1981.Interaction models for water in relation to protein hydration[J].Intermolecular Forces,14: 331-342 |
| [5] | Berendsen H J C,Postma J P M,van Gunsteren W F,et al.1984.Molecular dynamics with coupling to an external bath[J].The Journal of Chemical Physics,81(8): 3684-3690 |
| [6] | Brogren C,Karlsson H T.1997.A model for prediction of limestone dissolution in wet flue gas desulfurization applications[J].Industrial & Engineering Chemistry Research,36(9): 3889-3897 |
| [7] | Bruneval F,Donadio D,Parrinello M.2007.Molecular dynamics study of the solvation of calcium carbonate in water[J].The Journal of Physical Chemistry B,111(42): 12219-12227 |
| [8] | Chen Y J,Xu G Y.2013.Improvement of Ca2+-tolerance by the introduction of EO groups for the anionic surfactants: Molecular dynamics simulation[J].Colloids and Surfaces (A: Physicochemical and Engineering Aspects),424: 26-32 |
| [9] | Dang L X,Garrett B C.2004.Molecular mechanism of water and ammonia uptake by the liquid/vapor interface of water[J].Chemical Physics Letters,385(3/4): 309-313 |
| [10] | 丁红蕾.2010.氨基湿法烟气脱硫的机理及工业试验研究[D].杭州: 浙江大学.32-32 |
| [11] | Ebanks S C,O'Donnell M J,Grosell M.2010.Acquisition of Ca2+ and HCO3-/CO32- for shell formation in embryos of the common pond snail Lymnaea stagnalis[J].Journal of Comparative Physiology B,180(7): 953-965 |
| [12] | Garrett B C,Schenter G K,Morita A.2006.Molecular simulations of the transport of molecules across the liquid/vapor interface of water[J].Chemical Reviews,106(4): 1355-1374 |
| [13] | Hanus J,Mazeau K.2006.The xyloglucan-cellulose assembly at the atomic scale[J].Biopolymers,82(1): 59-73 |
| [14] | Himmelblau D M.1964.Diffusion of dissolved gases in liquids[J].Chemical Reviews,64(5): 527-550 |
| [15] | Karasawa N,Goddard W A III.1992.Force fields,structures,and properties of poly (vinylidene fluoride) crystals[J].Macromolecules,25(26): 7268-7281 |
| [16] | Kerisit S,Cooke D J,Spagnoli D,et al.2005.Molecular dynamics simulations of the interactions between water and inorganic solids[J].Journal of Materials Chemistry,15(14): 1454-1462 |
| [17] | Kiil S,Michelsen M L,Dam-Johansen K.1998.Experimental investigation and modeling of a wet flue gas desulfurization pilot plant[J].Industrial & Engineering Chemistry Research,37(7): 2792-2806 |
| [18] | 廖瑞金,贡春艳,周欣,等.2012.基于分子动力学模拟的油纸绝缘系统中气体小分子扩散行为[J].高电压技术,38(9): 2373-2382 |
| [19] | Mark P,Nilsson L.2001.Structure and dynamics of the TIP3P,SPC,and SPC/E water models at 298 K[J].The Journal of Physical Chemistry A,105(43): 9954-9960 |
| [20] | Matsumoto M.1996.Molecular dynamics simulation of interphase transport at liquid surfaces[J].Fluid Phase Equilibria,125(1/2): 195-203 |
| [21] | Moin S T,Lim L H V,Hofer T S,et al.2011.Sulfur dioxide in water: Structure and dynamics studied by an Ab initio quantum mechanical charge field molecular dynamics simulation[J].Inorganic Chemistry,50(8): 3379-3386 |
| [22] | Paul S,Chandra A.2005.Liquid-vapor interfacial properties of water-ammonia mixtures: Dependence on ammonia concentration[J].The Journal of Chemical Physics,123(17): 174712-1-174712-9 |
| [23] | Shamay E S,Johnson K E,Richmond G L.2011.Dancing on water: The choreography of sulfur dioxide adsorption to aqueous surfaces[J].The Journal of Physical Chemistry C,115(51): 25304-25314 |
| [24] | Somasundaram T,Lynden-Bell R M,Patterson C H.1999.The passage of gases through the liquid water/vapour interface: a simulation study[J].Physical Chemistry Chemical Physics,1(1): 143-148 |
| [25] | Souza S M A G U,Santos F B F,de Souza A A U,et al.2010.Limestone dissolution in flue gas desulfurization—experimental and numerical study[J].Journal of Chemical Technology and Biotechnology,85(9): 1208-1214 |
| [26] | Tarbuck T L,Richmond G L.2006.Adsorption and reaction of CO2 and SO2 at a water surface[J].Journal of the American Chemical Society,128(10): 3256-3267 |
| [27] | Vácha R,Slavíěek P,Mucha M,et al.2004.Adsorption of atmospherically relevant gases at the air/water interface: Free energy profiles of aqueous solvation of N2,O2,O3,OH,H2O,HO2,and H2O2[J].The Journal of Physical Chemistry A,108(52): 11573-11579 |
| [28] | Walbran S,Kornyshev A A.2001.Proton transport in polarizable water[J].The Journal of Chemical Physics,114(22): 10039 |
| [29] | Wang L D,Zhao Y.2008.Kinetics of sulfite oxidation in wet desulfurization with catalyst of organic acid[J].Chemical Engineering Journal,136(2/3): 221-226 |
| [30] | 王乃华,鲁天毅.2005.石灰石/石膏湿法烟气脱硫金属浆液循环泵国产化研究及实践[J].电力环境保护,21(2): 10-12 |
| [31] | Yang J,Ren Y,Tian A M,et al.2000.COMPASS force field for 14 inorganic molecules,He,Ne,Ar,Kr,Xe,H2,O2,N2,NO,CO,CO2,NO2,CS2,and SO2,in liquid phases[J].The Journal of Physical Chemistry B,104(20): 4951-4957 |
| [32] | Yuan R,Li Y,Li C X.2013.Study about how the metal cationic ions affect the properties of partially hydrolyzed hydrophobically modified polyacrylamide (HMHPAM) in aqueous solution[J].Colloids and Surfaces (A: Physicochemical and Engineering Aspects),434: 16-24 |
| [33] | 中国环境保护产业协会脱硫脱硝委员会.2013.我国脱硫脱硝行业2012年发展综述[J].中国环保产业,(7): 8-20 |
| [34] | 钟毅.2008.基于WFGD系统的硫、氮、汞污染物协同脱除的理论与实验研究[D].杭州: 浙江大学.58-126 |