 2014, Vol. 34
2014, Vol. 34



三氯乙烯(Trichloroethylene,TCE)作为氯代烃溶剂,在过去几十年中被广泛用于金属脱脂、喷涂、塑料制造等行业,并通过挥发、泄漏、不当处置等途径进入土壤、地下水和大气环境中,目前已成为环境中典型的有机污染物(Watts,1998).据美国一项污染场地调查结果表明,在美国受TCE污染的场地多达493个,约占氯代烃污染场地总数的69%;比利时某场地地下水中TCE的检出浓度高达25 mg · L-1,而地下水筛选值仅为70 μg · L-1(Crévecoeur et al., 2011),尤其是在污染严重的场地中,也曾多次发现TCE以纯自由相的形式存在于地下水或地下土壤中,成为土壤或地下水长期的污染源.TCE污染在我国部分城市的浅层地下水中也均有检出(张达政等,2002;何江涛等,2005).
原位化学氧化(ISCO)是污染场地修复治理的一项重要技术,其中,采用过硫酸盐氧化剂进行修复已引起人们广泛的关注.过硫酸钠作为一种强氧化剂,在工业上被大量用作漂白剂,其氧化还原电位E0达到2.01 V(House,1962).但过硫酸钠自身比较稳定,与大多数难以生物降解的有机污染物反应速率较慢(Block et al.,2004).然而,经紫外、超声、热、过渡金属等活化后,过硫酸钠会产生氧化还原电位更强的硫酸根自由基SO- ·4(E0≈2.6 V),其在理论上可降解绝大部分有机污染物.其中,过渡金属活化一直备受人们的关注,如Fe、Co、Mn、Ag等均已被证明具有活化能力,但与Fenton反应类似,过渡金属离子的活化反应一般需要在酸性条件下,并且活化速率过快(如Fe2+),降低了有机污染物的去除效率,在一定程度上限制了过渡金属活化的应用范围(Anipsitakis et al., 2004).而矿物活化则能克服上述缺点,磁铁矿(Fe3O4)、赤铁矿(Fe2O3)、硫化亚铁(FeS)和黄铁矿(FeS2)在降解有机污染物过程中被证明对H2O2和Na2S2O8有催化/活化能力(Matta et al., 2007;Che et al., 2011;Usman et al., 2012),如氯代烃、硝基苯、石油烃等.其中,磁铁矿在地下土壤环境中广泛分布,其作为一种含Fe2+和Fe3+混合价态的金属矿物,具有比单一Fe3+矿物更好的活化能力.Hou等(2012)研究了用超声优化磁铁矿活化过硫酸钠降解水溶液中的四环素,但以磁铁矿活化过硫酸盐来氧化降解氯代烃并将其应用在受污染泥浆体系中的研究尚未见报道.
因此,本研究采用磁铁矿(Fe3O4)活化过硫酸钠,以降解泥浆体系中的TCE,同时考察过硫酸钠在体系中的消耗,以及Fe2+、地下水中的阴离子和土壤成分中的有机质对TCE降解效果的影响,并观测反应过程中有机质的变化.
2 实验部分(Experimental) 2.1 实验材料TCE(纯度≥99.5%)、甲醇(纯度≥99.9%)、七水合硫酸亚铁、过硫酸钠、氯化钠、硝酸、浓硫酸、双氧水、重铬酸钾、碳酸氢钠、正己烷均为分析纯.磁铁矿(Fe3O4,≥97%),理化性质见表 1.比表面积以BET比表面积表征,晶体结构经X射线衍射分析(XRD),表面形态经电镜扫描分析(SEM),结果见图 1.经分析,磁铁矿纯度达到99%.
| 表1 磁铁矿理化性质 Table 1 Physico-chemical characteristics of magnetite |
 |
| 图 1 磁铁矿SEM和XRD图 Fig. 1 SEM and XRD images of magnetite |
实验土壤取自上海某氯代烃污染场地的2个位置,分为场地土S1和S2,取样深度为0~50 cm,将不同深度的土壤样品均匀混合后置于室内自然风干,拣去动、植物残体和石块等,研磨后经10目筛网(<0.2 mm)筛滤备用.将场地土S2进一步处理以去除土壤中的有机质,土壤有机质去除分别采用H2O2氧化处理和高温灼烧法处理,将土样反复加入质量分数为10%的H2O2并在机械搅拌下混合,直到加入H2O2不再产生明显气泡为止(张坤峰等,2010);采用马弗炉高温灼烧至恒重以完全去除土壤中的有机碳,冷却后同样经过风干、研磨和筛分处理,土壤理化性质见表 2.场地土溶出实验结果表明,两处场地土土样中TCE本底浓度均低于检出限(10 μg · L-1).
| 表2 土壤理化性质 Table 2 Physico-chemical characteristics of soils |
XW-80A涡旋振荡器,TDL-40B低速离心机,SX2-4-10箱式电阻炉,DELTA 320 pH计(Mettler-Toledo,Switzerl and ),UV-2100分光光度计,DF-101S集热式磁力加热搅拌器,7890A气相色谱仪(Agilent,USA),ASAP-2010N比表面孔径测定仪(Micromeritics,USA),X射线衍射仪(D/max 2550VB/PC,Janpan).
2.3 实验过程本研究采用批次试验,实验中土样与水质量比为1 ∶ 2,准确称取7.60 g土壤于20 mL螺纹口棕色瓶中,瓶盖内衬聚四氟乙烯/硅胶隔垫.配制一定浓度的TCE装满反应瓶,置于滚筒反应器中混合(6~7 r · min-1)24 h(Liang et al., 2003),试验开始前加入固态的过硫酸钠(0.60 g)和磁铁矿(0.20或0.40 g),到达反应时间后,取出1 mL 泥浆混合溶液,加入到含2 mL正己烷的萃取瓶中,取上部萃取液进行分析.根据10次平行回收率的测定实验结果(USEPA SW-846),该萃取方法TCE的回收率在85%~92%之间.将不投加过硫酸钠和磁铁矿的批次实验设为控制组实验,其余实验条件不变.实验中配制TCE浓度均为0.15 mmol · L-1(20 mg · L-1),除考察有机质变化实验外,Na2S2O8和磁铁矿浓度分别为126 mmol · L-1和为10 g · L-1,pH不调节,温度控制在(20±2)℃.
2.4 分析方法采用Agilent 7890A自动进样气相色谱仪测定TCE的含量.色谱柱型号为DB-VRX(长60 m、内径250 μm、厚度1.4 μm),检测器为ECD.TCE进样条件:进样口温度为240 ℃,柱温75 ℃,色谱柱流量5 mL · min-1,检测器温度为260 ℃,分流比20 ∶ 1,载气为N2.土壤总有机质(SOM)含量采用重铬酸钾外加热-容量法测定(GB/T50123—1999),检测限为0.01%~25.0%(宁梅,2010).泥浆中的过硫酸钠和铁离子浓度经过离心后移取上清液,分别采用碘化钾分光光度法(Liang et al., 2008)、邻菲啰啉法(Tamura et al., 1974)测定.
3 结果与讨论(Results and discussion) 3.1 过硫酸钠在磁铁矿体系中的分解过硫酸钠在各种反应体系中的变化结果如图 2所示.由图可知,7 d后,Na2S2O8在水溶液、泥浆、磁铁矿-泥浆体系中的分解率分别为9.9%、10.4%和31.6%.水溶液中Na2S2O8的分解源于自身的水解作用(House,1962)(式(1)).在泥浆体系中,Na2S2O8的分解率与在水溶液中近似,场地土样S2中总铁含量达到5830 mg · kg-1(表 2),但Na2S2O8分解率并未得到明显提升,说明土样中各类含铁矿物质未能与Na2S2O8发生明显的活化反应.而将磁铁矿加入到泥浆体系后Na2S2O8分解率得到了明显提升,表明其与过硫酸钠在泥浆体系中发生了反应.磁铁矿与过硫酸钠反应机理尚未系统建立,Fang等(2013)和晏井春等(2011)提出,过硫酸钠与磁铁矿的反应是表面活化反应,氧化剂与磁铁矿表面上的Fe2+直接反应产生SO- ·4自由基,SO- ·4经过一系列链式反应产生 · OH自由基,而在这一过程中未观察到表面的Fe2+/Fe3+转移到溶液中.这不同于其他含二价铁矿物活化氧化剂的反应机理,如黄铁矿(FeS2).Che等(2011)研究发现,黄铁矿催化H2O2是先由矿物表面的Fe2+转移到溶液中,释放出的Fe2+再与溶液中的H2O2反应产生SO- ·4.
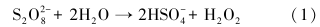
 |
| 图 2 Na2S2O8在各体系中的分解情况 Fig. 2 Na2S2O8 decomposition in various systems |
图 3中控制组实验结果表明,TCE在水溶液中或泥浆体系中经过48 h后均没有任何降解趋势,但在过硫酸钠存在的条件下,TCE降解明显,同时TCE降解符合准一级反应动力学方程(动力学参数见表 3).经过48 h后,只加入过硫酸钠的泥浆体系中TCE去除率达到了50.1%,而加入磁铁矿(10 g · L-1 和20 g · L-1)体系的TCE去除率则分别达到了83.3%和97.0%,反应速率常数分别为0.0359 h-1和0.0610 h-1.由于土壤中含有大量的缓冲物质(如各种弱酸、腐殖酸及其盐类等),导致加入过硫酸钠后泥浆系统的pH值偏中性分别为7.38(10 g · L-1)和7.39(20 g · L-1),而Na2S2O8自身经过水解或活化产物SO- ·4,均会生成HSO-4,释放出H+(式(1)~(3)),使整个反应体系pH值略微降低至7.00(10和20 g · L-1).可见48 h反应结束后,pH值仍接近中性,减小幅度在0.24~0.39之间.

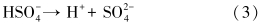
 |
| 图 3 TCE在磁铁矿-泥浆体系中的降解行为 Fig. 3 TCE degradation in magnetite-soil slurry system |
| 表3 各种反应条件下的实验参数 Table 3 Parameters under various experimental conditions |
反应过程中含磁铁矿(10 g · L-1)的泥浆混合液离心后上清液中总铁浓度为9.72 mg · L-1,仅占加入磁铁矿中总铁比例的0.14%,反而低于纯泥浆体系中所测得的总铁浓度(9.86 mg · L-1),溶液中的总铁可能来源于土壤中可溶性铁.上述结果表明,过硫酸钠的投入并未使磁铁矿中的铁释放到溶液中,该结论与之前应用磁铁矿活化过硫酸钠文献中的结果一致(Fang et al., 2013;Hou et al., 2012).推测这可能是由磁铁矿结构性质决定的,铁离子与过硫酸钠之间的反应只在磁铁矿表面进行,Fe2+和Fe3+并未从固体颗粒表面转移到溶液中,反应后磁铁矿表面的变化经由XRD检测发现,衍射角在28.5°和60.8°位置上出现了α-Fe2O3的对应峰(图 4),证明了其是磁铁矿与过硫酸钠反应后在表面形成的主要产物.
 |
| 图 4 磁铁矿变化XRD图(a.反应前磁铁矿;b.反应后磁铁矿) Fig. 4 XRD image of the magnetite change(a.original magnetite; b.spent magnetite) |
Fe2+普遍存在于地下水及土壤环境中,可能对TCE的降解过程产生重要影响.本文选择将0.1、1、10 mmol · L-1 Fe2+分别投加到含有126 mmol · L-1 Na2S2O8和10 g · L-1磁铁矿的泥浆体系中进行试验,结果如图 5所示.由图可知,当Fe2+浓度为0.1 mmol · L-1时,TCE去除率(82.4%)与不添加Fe2+的实验结果(83.3%)近似.这可能是由于Fe2+浓度过低,活化过硫酸钠后产生的SO- ·4有限,同时,Fe2+与过硫酸钠反应速率常数k值为2×101 mol · L-1 · s-1(Travina et al., 1999),因此,一旦加入泥浆中,泥浆成分迅速会对有限的SO- ·4产生竞争,未对TCE的去除效率产生影响.而当Fe2+浓度明显增大后,TCE的去除率明显提升,反应速率常数与Fe2+浓度成正比(表 3).Fe2+投加量为1、10 mmol · L-1时,速率常数分别增大到0.0468 h-1和0.0900 h-1,TCE的最终去除率均为100%.反应速率加快的主要原因是:①投加的Fe2+会吸附在磁铁矿表面,增大电子密度,降低矿物表面与Fe2+关联时的氧化还原电位,有助于电子转移到污染物分子,从而强化TCE的还原脱氯反应(Amonette et al., 2000;Gregory et al., 2004);②当氧化剂存在时,吸附在磁铁矿表面的Fe2+会活化Na2S2O8,并产生具有更强氧化能力的硫酸根自由基(SO- ·4)(式(4)),由此加快了TCE降解速率(House,1962).
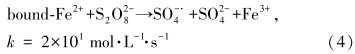
 |
| 图 5 Fe2+对TCE降解的影响 Fig. 5 Effect of Fe2+ ion on TCE degradation |
土壤和地下水的状况,特别是阴离子的存在对目标污染物的降解会产生一定的影响.因此,本研究选择土壤和地下水中普遍存在的Cl-和HCO-3,分别考察了1、10和100 mmol · L-1浓度下TCE的降解情况.由图 5可知,Cl- 浓度为1、10 mmol · L-1时,TCE在48 h后的去除率分别为80.6%和79.4%,对TCE降解影响较小;而在100 mmol · L-1时,反应速率明显降低,去除率为70.4%(表 3).Cl-超过一定浓度后对TCE降解产生抑制作用的主要原因可能是:高浓度的Cl- 与SO- ·4反应产生低活性的氯自由基(Criquet et al., 2009)(式(5)~(8)),虽然有文献(Khan et al., 2010)报道生成的氯自由基会增加体系中总自由基的数量而有利于污染物降解,但由于本实验中采用的目标污染物TCE的难降解性,低活性的氯自由基并不能充分分解 TCE,因此,Cl-的加入仅起到了消耗体系中SO- ·4的作用.HCO-3作为土壤中起主要缓冲作用的物质,含量一般为24.4 mg · kg-1,同时HCO-3也是较强的自由基清扫剂(式(8)),因此,其对TCE的降解影响较Cl-更为显著.当 HCO-3投加量为 1、10、100 mmol · L-1时,TCE在反应48 h后的去除率分别降至77.4%、63.8%、50.1%(图 6).
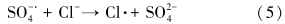
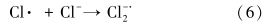
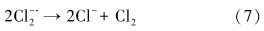

 |
| 图 6 Cl-和HCO-3对TCE降解的影响 Fig. 6 Effects of Cl- and HCO-3 anions on TCE degradation |
除阴离子外,土壤有机质(Soil Organic Matter,SOM)也是影响TCE在泥浆体系中降解速率的重要因素.实验中4种土样的SOM含量由大到小顺序为:场地土S1>场地土S2>H2O2处理场地土S2>高温灼烧处理场地土S2(表 2),可见随着有机质含量的减少,TCE降解效果则变好(图 7),TCE的降解速率与SOM含量大小成反比(表 3).土样经H2O2处理后去除了SOM中的低聚合的软碳(Soft Carbon)部分(张坤峰等,2010;Cuypers et al., 2000);经800 ℃灼烧后,去除了全部的有机质(包含软碳和硬碳).两种方法处理后SOM分别降为0.32%和0,TCE反应速率则由原土的0.0359 h-1增大到0.0542 h-1和0.0921 h-1,去除率均达到100%.造成这种有机质对TCE降解效果影响的主要原因为:①土壤中SOM会消耗部分Na2S2O8及其产生的氧化性自由基.有研究表明,在高温条件下(80~90 ℃/40~60 ℃),Na2S2O8与有机质之间存在着较强的反应活性(Doyle et al., 2004;Siregar et al.,2005;Peyton,1993),但Hønning等(2007)在常温下测得10 d内过硫酸钠消耗量仅为0.08~0.24 g · kg-1,KMnO4为1~8 g · kg-1.这是由于S2O2-8在常温条件下对土壤有机质的亲和性小于KMnO4与H2O2,选择性地氧化了软碳部分.②经高温灼烧后,泥浆的pH变化范围为11.3~11.6,pH值上升的原因可能是源于高温破坏了土样中的缓冲物质,并使某些矿物质分解产生碱性物质(如CaCO3分解为CaO).在pH>11时,OH-会促进Na2S2O8的分解产生比SO- ·4氧化还原电位更高的OH ·(式(9))(Furman et al., 2010;House,1962).
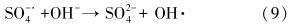
 |
| 图 7 有机质含量对TCE降解效果的影响 Fig. 7 Effect of SOM content on TCE degradation |
土壤有机质含量的变化可间接体现反应过程中氧化剂的消耗量.为进一步考察磁铁矿活化Na2S2O8时SOM含量的变化,本研究中选择了63、126、252 mmol · L-1 3种浓度的Na2S2O8,磁铁矿投加量分别为10 g · L-1和20 g · L-1,以考察反应过程中土壤有机质的变化,结果如图 8所示.由图 8a可知,经过48 h反应后,磁铁矿为10 g · L-1条件下,氧化剂浓度为252 mmol · L-1时SOM的变化较63 mmol · L-1和126 mmol · L-1时大,减少了1.27 g · kg-1.含20 g · L-1磁铁矿的实验组中,反应48 h后SOM的变化较10 g · L-1实验组稍大,也仅降低了1.63 g · kg-1.同时如图 8所示,SOM变化趋势呈现不规律性,结果与Kuei-Jyum Yeh等(2002)实验中采用Fenton试剂氧化土壤中氯酚的反应过程中SOM变化也非单一趋势相似.推测造成这种现象的原因可能是受试土样SOM分布的非均相性和目前检测SOM方法灵敏度较低.实验结果显示,在常温条件下,反应中SOM对TCE降解的影响更接近于Hønning等(2007)的结论,即SOM对TCE降解的抑制并非单纯是SOM与Na2S2O8及其产生的自由基发生反应,但考虑到实验条件的差异,不能排除SOM仍会与TCE竞争过硫酸钠及其产生的自由基.SOM对TCE降解的抑制可能是通过消耗过硫酸钠、表面消耗自由基和对有机污染物的竞争吸附等多种途径引起的(Miller et al., 1999;Costanza et al., 2010).
 |
| 图 8 反应过程中有机质变化(a.10 g · L-1磁铁矿;b.20 g · L-1磁铁矿) Fig. 8 SOM changes in reaction(a.10 g · L-1 magnetite; b.20 g · L-1 magnetite) |
1)在非原位修复实验中,磁铁矿活化过硫酸钠技术能有效去除泥浆体系中的TCE,过硫酸钠浓度为126 mmol · L-1,磁铁矿投加量为20 g · L-1时,48 h内TCE去除率达到97%,且TCE降解过程符合准一级反应动力学过程.
2)投加Fe2+(1~10 mmol · L-1)对磁铁矿活化过硫酸钠降解泥浆体系中的TCE起到促进作用,Cl-(10~100 mmol · L-1)和HCO-3(1~100 mmol · L-1)对磁铁矿活化过硫酸钠降解泥浆体系中的TCE起抑制作用,HCO-3的抑制效果尤为显著.
3)土壤有机质对TCE的降解影响显著,高有机质含量会抑制TCE的降解.反应过程中SOM的减少并不显著,表明SOM并非单纯消耗氧化剂,而是通过消耗过硫酸钠、表面消耗自由基和对有机污染物的竞争吸附等多种途径共同影响TCE的降解.
| [1] | Amonette J E,Workman D J,Kennedy D W,et al.2000.Dechlorination of carbon tetrachloride by Fe (Ⅱ) associated with goethite [J].Environmental Science & Technology,34(21):4606-4613 |
| [2] | Anipsitakis G P,Dionysiou D D.2004.Radical generation by the interaction of transition metals with common oxidants[J].Environmental Science & Technology,38(13):3705-3712 |
| [3] | Block P A,Brown R A,Robinson D.2004.Novel activation technologies for sodium persulfate in situ chemical oxidation//Proceedings of the Fourth International Conference on the remediation of chlorinated and recalcitrant compounds.Monterey,CA:Battelle Press |
| [4] | Che H,Bae S,Lee W.2011.Degradation of trichloroethylene by Fenton reaction in pyrite suspension[J].Journal of Hazardous Materials,185(2/3):1355-1361 |
| [5] | Costanza J,Otaño G,Callaghan J,et al.2010.PCE oxidation by sodium persulfate in the presence of solids[J].Environmental Science & Technology,44(24):9445-9450 |
| [6] | Crévecoeur S,Debacker V,Joaquim-Justo C,et al.2011.Groundwater quality assessment of one former industrial site in Belgium using a TRIAD-like approach[J].Environmental Pollution,159(10):2461-2466 |
| [7] | Criquet J,Leitner N K V.2009.Degradation of acetic acid with sulfate radical generated by persulfate ions photolysis[J].Chemosphere,77(2):194-200 |
| [8] | Cuypers C,Grotenhuis T,Joziasse J,et al.2000.Rapid persulfate oxidation predicts PAH bioavailability in soils and sediments[J].Environmental Science & Technology,34(10):2057-2063 |
| [9] | Doyle A,Weintraub M N,Schimel J P.2004.Persulfate digestion and simultaneous colorimetric analysis of carbon and nitrogen in soil extracts[J].Soil Science Society of America Journal,68(2):669-676 |
| [10] | Fang G D,Dionysiou D D,Al-Abed S R,et al.2013.Superoxide radical driving the activation of persulfate by magnetite nanoparticles:Implications for the degradation of PCBs[J].Applied Catalysis (B:Environmental),129:325-332 |
| [11] | Furman O S,Teel A L,Watts R J.2010.Mechanism of base activation of persulfate[J].Environmental Science & Technology,44(16):6423-6428 |
| [12] | Gregory K B,Larese-Casanova P,Parkin G F,et al.2004.Abiotic transformation of hexahydro-1,3,5-trinitro-1,3,5-triazine by FeⅡ bound to magnetite[J].Environmental Science & Technology,38(5):1408-1414 |
| [13] | 何江涛,李烨,刘石,等.2005.浅层地下水氯代烃污染的天然生物降解[J].环境科学,26(2):121-125 |
| [14] | Hønning J,Broholm M M,Bjerg P L.2007.Quantification of potassium permanganate consumption and PCE oxidation in subsurface materials[J].Journal of Contaminant Hydrology,90(3/4):221-239 |
| [15] | Hou L W,Zhang H,Xue X F.2012.Ultrasound enhanced heterogeneous activation of peroxydisulfate by magnetite catalyst for the degradation of tetracycline in water[J].Separation and Purification Technology,84:147-152 |
| [16] | House D A.1962.Kinetics and mechanism of oxidations by peroxydisulfate[J].Chemical Reviews,62(3):185-203 |
| [17] | Khan N E,Adewuyi Y G.2010.Absorption and oxidation of nitric oxide(NO) by aqueous solutions of sodium persulfate in a bubble column reactor[J].Industrial & Engineering Chemistry Research,49(18):8749-8760 |
| [18] | Kuei-Jyum Yeh C,Kao Y A,Cheng C P.2002.Oxidation of chlorophenols in soil at natural pH by catalyzed hydrogen peroxide:the effect of soil organic matter[J].Chemosphere,46(1):67-73 |
| [19] | Liang C J,Bruell C J,Marley M C,et al.2003.Thermally activated persulfate oxidation of trichloroethylene (TCE) and 1,1,1-trichloroethane (TCA) in aqueous systems and soil slurries[J].Soil and Sediment Contamination:An International Journal,12(2):207-228 |
| [20] | Liang C J,Huang C F,Mohanty N,et al.2008.A rapid spectrophotometric determination of persulfate anion in ISCO[J].Chemosphere,73(9):1540-1543 |
| [21] | Matta R,Hanna K,Chiron S.2007.Fenton-like oxidation of 2,4,6-trinitrotoluene using different iron minerals[J].Science of the Total Environment,385(1):242-251 |
| [22] | Miller C M,Valentine R L.1999.Mechanistic studies of surface catalyzed H2O2 decomposition and contaminant degradation in the presence of sand[J].Water Research,33(12):2805-2816 |
| [23] | 宁梅.2010.土壤有机质含量测定方法研究.辽宁省环境科学学会2010年学术年会及"发展辽宁生态工业建设低碳经济社会"论坛.沈阳.130-133 |
| [24] | Peyton G R.1993.The free-radical chemistry of persulfate-based total organic carbon analyzers[J].Marine Chemistry,41(1/3):91-103 |
| [25] | Siregar A,Kleber M,Mikutta R,et al.2005.Sodium hypochlorite oxidation reduces soil organic matter concentrations without affecting inorganic soil constituents[J].European Journal of Soil Science,56(4):481-490 |
| [26] | Tamura H,Goto K,Yotsuyanagi T,et al.1974.Spectrophotometric determination of iron(Ⅱ) with 1,10-phenanthroline in the presence of large amounts of iron(Ⅲ)[J].Talanta,21(4):314-318 |
| [27] | Travina O A,Kozlov Y N,Purmal A P,et al.1999.Synergism of the action of the sulfite oxidation initiators,iron and peroxydisulfate ions[J].Russian Journal of Physical Chemistry,73(8):1215-1219 |
| [28] | Usman M,Faure P,Hanna K,et al.2012.Application of magnetite catalyzed chemical oxidation (Fenton-like and persulfate) for the remediation of oil hydrocarbon contamination[J].Fuel,96:270-276 |
| [29] | Watts R J.1998.Hazardous Wastes:Sources,Pathways,Receptors[M].New York:John Wiley & Sons |
| [30] | 晏井春,朱丽华,陆晓华,等.2011.纳米四氧化三铁活化过硫酸盐降解RhB的研究//第六届全国环境化学大会暨环境科学仪器与分析仪器展览会摘要集.北京:中国化学会 |
| [31] | 张达政,陈鸿汉,李海明,等.2002.浅层地下水卤代烃污染初步研究[J].中国地质,29(3):326-329 |
| [32] | 张坤峰,何江涛,刘明亮,等.2010.土壤有机质形态对有机污染物三氯乙烯(TCE)的吸附影响研究[J].安徽农业科学,38(1):286-288 |