 2014, Vol. 34
2014, Vol. 34



全氟化合物(Perfluorinated compounds,PFCs)中的全氟辛烷磺酸(Perfluorooctanesulfonate,PFOS)和全氟辛酸(Perfluorooctanoate,PFOA)具有疏水和疏油特性及良好的表面活性和耐热性,因而在全球范围内被用于工业产品及生活用品的生产中,例如,纺织品的表面保护材料、泡沫灭火剂、感光材料表面处理剂、半导体工业清洁和表面处理液、地板蜡、杀虫剂、航空液压油、防水和防油纸等(OECD,2002).PFOS和PFOA不易在环境中代谢和降解,具有生物累积作用,在生物及相关工业工人的血液和组织样品中均有检出(Calafat et al., 2006a;2006b;Kannan et al., 2004).PFOS和PFOA亦具有内分泌干扰活性和致癌性(Jensen et al., 2008;Butenhof et al., 2012),因此,在2009年5月《关于持久性有机物的斯德哥尔摩公约(POPs公约)》第四次缔约方大会上,将PFOS及其盐类和全氟辛基磺酰氟列入公约附件B(限制生产、使用和进出口).
欧美(Bossi et al., 2008;Huset et al., 2008;Ahrens et al., 2011)、日本(Takazawa et al., 2009)、韩国(Guo et al., 2010)和中国(Sun et al., 2011)等许多国家有关污水处理厂PFCs的报道表明,城市污水处理厂排水是PFCs进入水环境的主要途径之一.2006年,Skutlarek等(2006)报道了德国由于未对含PFCs废水进行合理处置,导致了河流的严重污染,在一些区域甚至导致了饮用水的污染.污水中的PFOS和PFOA主要来自经过表面处理的产品(如衣物和地毯)的清洗和护理,含PFCs的化妆品及工业产品的浸出(Boulanger et al., 2005).由于消费者使用了含有PFCs的产品,所以这些物质被发现存在于市政污水(Schultz et al., 2006;Sinclair et al., 2006)和污泥(Higgins et al., 2005)中.PFOS和PFOA不易在污水处理过程中被降解,进而分配于水相和污泥中.并且,大多数的研究表明,经传统的生物处理后,污水中的PFOA和PFOS浓度反而会增加(Schultz et al., 2006).来自污水处理厂的PFCs会通过污水出水、化粪池出水和污泥还田等途径进入环境中从而造成污染.已有研究对PFCs在污水处理厂污水和污泥中的分布进行了报道,但对于在污水处理整个流程的每一步处理工艺中,PFCs在水相和泥相间的分布和迁移转化规律还了解甚少.污水处理厂是PFCs进入环境中的污染源之一,研究PFCs在污水处理厂中的迁移转化规律有助于管理和控制PFCs通过这一途径进入环境.目前,中国污水处理厂中PFCs存在状况的研究刚刚起步,鲜有相关报道,污染情况的调查研究工作还较为缺乏.随着近年来中国对化学品管理力度的加强,以及对《POPs公约》的履约要求,有必要对新增POPs物质的污染源进行基础调查.
脱氮除磷污水处理工艺是目前北京等城市污水厂采用的主要工艺,本研究针对北京市卢沟桥污水处理厂倒置A2O工艺流程各阶段的污水和污泥进行采样和浓度分析检测,分析PFOS和PFOA在好氧、缺氧和厌氧不同条件下污水和污泥中的浓度分布特征,以及泥水分配规律.同时,计算并解释了各工艺阶段PFOS和PFOA的物质流的变化,以期为PFOS和PFOA在污水处理厂的处理情况提供参考,并为环境中PFOS和PFOA的来源提供基础数据.
2 材料与方法(Materials and methods) 2.1 试剂与材料PFOS和PFOA购自美国Sigma-Aldrich公司;GF/F玻璃纤维滤膜(0.47 μm)购自美国Whatman公司;乙酸(99.5%)、氨水(25.0%~28.0%)、乙酸铵(98.0%)购自国药集团化学试剂有限公司;色谱级甲醇和二氯甲烷购自美国J.T.Baker公司;WAX固相萃取小柱(150 mg,6 cm3)、Sep-Pak Silica固相萃取小柱(1 g,6 cm3)和Sep-Pak Plus型水样富集器均购自美国Waters公司;超纯水由Milli-Q synthesis超纯水机(Millipore,美国)制备.
2.2 采样选择北京市卢沟桥污水处理厂为调查对象,该厂日处理污水量冬季约为70000 m3 · d-1,夏季约为90000 m3 · d-1,服务区覆盖北京南部人口约364000人,污水来源主要为生活和商业污水.污水处理工艺由预处理装置、初级处理装置和二级生物处理装置构成,具体如图 1所示.预处理装置包括粗格栅和细格栅,初级处理装置为圆形沉淀池,二级生物处理使用改良倒置式A2O工艺,由缺氧1区-厌氧区-好氧1区-缺氧2区-好氧2区组成.污水40%进入缺氧工段,60%进入厌氧工段,汇合后再经过好氧、缺氧和好氧处理后,进入二次沉淀池,大部分出水排放到附近的马草河中,另外约10000 m3 · d-1的出水经再生水厂处理后作为中水回用.二沉池的污泥90%经回流进入缺氧池1,其余作为剩余污泥排出,脱水后制成泥饼外运.
样品采集时间为2012年7月27日和11月30日,采样点布设如图 1所示.污水及泥水混合物收集于1.5 L的PET塑料瓶中,放置在有冰板的冷藏箱中运回实验室,在当天进行水样的过滤及前处理.进水和出水经GF/F玻璃纤维滤膜(0.47 μm)过滤处理,其余泥水混合物静置离心后进行泥水分离.水相在24 h内进行前处理,进出水的悬浮颗粒物及其余采样点的污泥进行冷冻干燥.
 |
| 图 1 污水处理厂工艺流程示意图及采样点(采样点S1~S8分别为进水、缺氧1、厌氧、好氧1、缺氧2和好氧2出水、二沉池回流污泥和出水) Fig. 1 Sketch map of wastewater treatment process and sampling sites.(Sampling sites S1~S8 are located at influent,anoxic zone 1,anaerobic zone,aerobic zone 1,anoxic zone 2,aerobic zone 2,returned sludge and effluent) |
将500 mL污水样品经GF/F玻璃纤维滤膜(0.47 μm)过滤后,采用英国标准协会BS ISO 25101(BSI,2009)和Yu等(2009)报道的固相萃取法进行水样的前处理.首先,WAX固相萃取柱分别用4 mL的0.1%氨水/甲醇溶液、4 mL甲醇和4 mL纯水依次活化(流速约3 mL · min-1);然后将水样减压通过固相萃取柱,富集体积均为0.5 L,控制流速3 mL · min-1.富集完成后,用4 mL的0.025 mol · L-1(pH=4)的乙酸盐缓冲溶液(乙酸+乙酸铵溶液)淋洗,将淋洗后的WAX固相萃取柱冷冻干燥3 h.之后再用4 mL的0.1%氨水/甲醇溶液洗脱,洗脱液收集于10 mL的聚丙烯管中,并将洗脱液用6 mL二氯甲烷稀释到10 mL,待用Silica固相萃取小柱进行净化.Sep-Pak Silica固相萃取小柱经5 mL二氯甲烷/甲醇(V/V=3 ∶ 2)活化后,净化稀释后的洗脱液(10 mL),控制流速3 mL · min-1,并用玻璃离心管接液体.将洗脱液用氮吹仪吹干,用甲醇定容至0.5 mL,涡旋混匀后转移至装有内衬管的进样瓶中,进行LC/MS/MS分析.
将冷冻后的污泥样品置于冷冻干燥机(Christ Alpha 1-4 LD plus,John Morris Scientific公司,澳大利亚)(-30 ℃以下)内干燥后,采用Higgings等(2005)使用的方法进行污染物提取和净化.首先,将100 mg样品放入容量为50 mL的聚丙烯离心管A中,加入7.5 mL的1%乙酸后涡旋混合1 min,超声水浴15 min(60 ℃).之后放入离心机中离心2 min,转速为3000 r · min-1,将提取的上清液加入另一个50 mL聚丙烯离心管B.再向A中加入2.5 mL的甲醇/1%乙酸(90/10,体积比)溶液,重复上述提取过程.再用相同量的乙酸溶液和甲醇/乙酸溶液重复两次上述提取过程,并将上清液转移至管B中.最后将管A置于离心机中离心2 min,转速为3000 r · min-1,并收集上清液于管B,收集于管B中的上清液总共约30 mL.将管B中的上清液进行WAX固相萃取及净化,步骤同污水样品.最后用0.5 mL甲醇定容,涡旋混合均匀后转移至装有内衬管的进样瓶中,进行LC-MS/MS分析.500 mL的进水和出水样品(S1和S8)过滤后载有悬浮固体SS的滤膜经过与污泥类似的提取净化过程后,用0.5 mL甲醇定容,涡旋混合均匀后过0.22 μm的尼龙滤头,然后转移至装有内衬管的进样瓶中,进行LC-MS/MS分析.
2.4 仪器分析仪器分析采用高效液相色谱和三重四极杆串联质谱联用系统(LC-MS/MS,API3200,Applied Biosystems公司,美国).色谱和质谱条件参考了Lin等(2012)的报道.
色谱条件:色谱柱为Ultimate C18柱(4.6 mm×250 mm,5 μm,Welch Mareials公司,美国);流动相A:甲醇,流动相B:5 mmol · L-1乙酸铵水溶液;流速:0.8 mL · min-1,柱温40 ℃,进样量10 μL,采用梯度洗脱方式,洗脱程序:0~2 min,流动相A从70%线性变化到95%;2~5 min,流动相A保持在95%;5~5.1 min,流动相A从95%线性变化到70%;5.1~7 min,流动相A保持在75%.
质谱条件:采用电喷雾离子源ESI负离子模式,碰撞气(CAD)0.034 MPa,气帘气(CUR)0.17 MPa,离子源电压(IS)-4500 V,离子源温度(TEM)550 ℃.选择性PFOS和PFOA监测离子质荷比(m/z)为499>80和413>369的带负电准分子离子.
2.5 标准曲线、回收率和空白实验用甲醇/水(V/V=1 ∶ 1)溶液配制浓度范围为5~200 μg · L-1的PFOS和PFOA混合标准液,测定标准液浓度,绘制标准曲线,线性可决系数>0.99.在过滤后的进水和回流污泥样品中分别添加100 ng · L-1和250 ng · g-1的PFOS/PFOA混合标准溶液,按照2.3节所述的前处理步骤进行富集,进水和回流污泥分别设3个平行样,根据测定结果计算加标回收率,结果如表 1所示.以信噪比为3确定仪器检测限(LOD),PFOS和PFOA的检测限分别为0.6 ng · L-1和0.1 ng · L-1.空白实验使用Milli-Q超纯水进行,超纯水(3个平行样)经过与污水同样的前处理步骤后,使用LC-MS/MS进行分析检测,PFOS和PFOA均低于检出限,所以前处理步骤没有引入污染.
| 表1 污水和污泥中PFOS和PFOA的LOD和回收率 Table 1 LOD and recovery of PFOS and PFOA in wastewater and sludge |
如图 2所示,进水中PFOS和PFOA的浓度范围分别为113.9~160.6 ng · L-1和14.7~68.1 ng · L-1,其中,夏季分别为(115.7±1.7)ng · L-1和(15.3±0.6)ng · L-1,冬季分别为(159.4±1.2)ng · L-1和(68.8±0.7)ng · L-1.文献报道中污水处理厂进出水PFOS和PFOA的浓度如表 2所示,相比较后发现,卢沟桥污水处理厂进水中PFOS的浓度与文献中其他污水处理厂在同一数量级,但PFOA的浓度仅与Murakami等(2009)报道的日本关东地区,以及Lin等(2010)报道的中国台湾的污水处理厂中PFOA的浓度类似,与其他研究相比较低.从图 2中可以明显看出,污水中PFOS浓度高于PFOA.这与美国肯塔基(Loganathan et al., 2007)、美国纽约(Sinclair et al., 2006)、德国(Busch et al., 2010)、韩国(Kim et al., 2012)、新加坡(Yu et al., 2009)及中国上海和大连两座工业污水厂(Chen et al., 2012)的情况相反,然而,这些报道中的污水处理厂的进水都含有工业来源或商业来源,且比例较大.对于进水来源主要是市政污水的污水处理厂而言,如表 2所列出的瑞士(Huset et al., 2008)、日本(Murakami et al., 2009)、中国台湾(Lin et al., 2010)和中国沿海城市(Chen et al., 2012)与本研究类似,进水中PFOS的浓度高于PFOA的浓度.可见,PFOS较PFOA在市政污水中存在更多,而PFOA受工业污水和商业污 水的影响较大.Chen等(2012)通过对中国12座污水处理厂的调查发现,工业污水来源占70%的污水处理厂中PFOS和PFOA的浓度分别为2.6~9.5 ng · L-1和47000~66000 ng · L-1,而市政污水进水中PFOS和PFOA的浓度分别为1.8~176.0 ng · L-1和2.6~191.0 ng · L-1.所以,Chen等推测,市政污水是PFOS的重要来源,而相当一部分的PFOA源自工业.其他的研究也说明了市政污水是城市污水处理厂中PFOS的持续性来源(Huset et al., 2008;Sinclair et al., 2006;Schultz et al., 2006;Bossi et al., 2008).而在没有工业废水进入的城市污水处理厂中,PFOA主要来自商业用途,如医院、商场和办公大楼中对经过全氟化合物进行表面处理后物品的清洗(Sinclair et al., 2006;Lin et al., 2010).卢沟桥污水处理厂的进水主要为市政污水,所以,PFOS浓度较PFOA高的情况与现有研究一致.冬季污水中PFOS和PFOA的浓度比夏季略高,但整体差别不大,且污水中的PFOS和PFOA的浓度在工艺流程的各阶段的变化规律在夏季和冬季也较为类似.
 |
| 图 2 PFOS和PFOA在夏季和冬季各工艺阶段污水、污泥和SS中的浓度分布 Fig. 2 Concentrations of PFOS and PFOA in water and sludge of each treatment process stage in summer and winter |
| 表2 污水处理厂污水和污泥中的PFOS和PFOA的浓度 Table 2 Concentrations of PFOS and PFOA in wastewater and sludge in previous studies |
如图 2所示,污泥中PFOS和PFOA的含量范围分别为110.4~669.5 ng · g-1和20.2~178.2 ng · g-1,其中,夏季分别为93.4~128.9 ng · g-1和20.2~55.5 ng · g-1,冬季分别为132.6~669.5 ng · g-1和70.0~178.2 ng · g-1.表 2中列出了文献报道的活性污泥中PFOS和PFOA的含量,本研究的活性污泥中PFOS的含量与Yan等(2012)报道的上海污水处理厂,以及Kim等(2012)报道的韩国的污水处理厂中的含量相似,比Chen等(2012)所报道的中国沿海城市污水处理厂污泥中PFOS的含量高.PFOA的含量相对PFOS较低,与Higgins等(2005)报道的美国旧金山的污水处理厂污泥中含量(n.d~29.4 ng · g-1)类似.污泥中PFOA的含量特征在夏季和冬季较为类似,但污泥中PFOS的含量特征在夏季和冬季有着明显的不同.夏季污泥中PFOS的含量在各工艺阶段差别不大,在冬季,两个好氧池中PFOS的含量有明显的升高(图 2).研究表明,污水中有许多前驱物质如N-EtFOSE(2(N-ethyl-perfluorooctane sulfonamido)ethyl alcohol)、N-EtFOSAA(2-(N-ethylperfluorooctane sulfonamido)acetic acid)、N-MeFOSAA(2-(N-methylperfluorooctane sulfonamido)acetic acid)可以分解产生PFOS(Higgins et al., 2005;Boulanger et al., 2005).所以,冬季厌氧和缺氧1(S2和S3)中PFOS的含量升高,在曝气的条件下,好氧池中的污泥对PFOS的吸附能力较大,使得在两个好氧池(S4和S6)的污泥中PFOS的含量增大.另外,因为冬季污泥停留时间(22 d)比夏季(18 d)长,不易被生物降解的PFOS会在污泥中积累,所以产生了冬季PFOS在各流程之间的变化幅度大于夏季的现象,而PFOA却没有这么明显的变化,这与两种物质的泥水分配规律不同有关(在3.2节中进行探讨).另外,采样方法对浓度分布特征也有较为明显的影响,由于本研究采用抓取的采样方法,污泥样品可能为泥龄较大的污泥,所以,出现了在S4和S6冬季的污泥中PFOS含量增高较为明显的现象.
由于进水和出水中不存在污泥相,所以依据2.3节中所述的前处理方法分析了进水和出水样品(S1和S8)悬浮固体SS中PFOS和PFOA的浓度,表示为每升污水的SS中PFOS和PFOA的浓度,分别为20.6~22.2 ng · L-1和10.4~11.2 ng · L-1(图 2,此处SS的单位为ng · L-1而非ng · g-1,每升污水的SS中的浓度与水相的浓度直接相加即可得出进水和出水中PFOS和PFOA的综合浓度;对于含有污泥相的S2~S7,综合浓度=水相浓度+泥相浓度×MLSS).
经过处理后,出水中PFOS和PFOA的浓度范围分别为60.1~116.3 ng · L-1和29.9~71.5 ng · L-1,其中,夏季分别为(65.8±5.7)ng · L-1和(30.1±0.2)ng · L-1,冬季分别为(157.9±74.7)ng · L-1和(70.0±1.5)ng · L-1.从图 2中可以看出,PFOA在出水中的浓度反而升高,这是由于污水中亦存在可能分解产生PFOA的前驱物质(Fluorotelomer alcohols)(Wang et al., 2005),从而使出水浓度升高.PFOS较PFOA而言,更易受污泥吸附的影响(Yu et al., 2009;Kunacheva et al., 2011),所以部分随剩余污泥排出或随回流污泥进入缺氧池1中,导致出水中PFOS的浓度有轻微下降.经污水处理厂处理后的PFOS和PFOA在污水和污泥中的总浓度都有所升高,这与PFOS和PFOA不能被生物降解有关.
3.2 PFOS和PFOA的泥水分配特征PFOS和PFOA在污水和污泥之间的分配用分配系数Kd来表示,可以体现不同条件下污染物在泥水之间的迁移转化规律及对去除的影响.Kd用如下公式来进行计算:
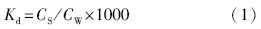
PFOS和PFOA在污水处理厂不同工艺阶段污水和污泥中的分配系数如表 3所示,PFOS和PFOA的Kd值的范围分别为894~11824 L · kg-1和544~2569 L · kg-1.略高于Zhou等(2010)报道的200~4050 L · kg-1和150~300 L · kg-1,Yu等(2009)报道的720~2324 L · kg-1和188~597 L · kg-1,以及Arvaniti等(2012)报道的398~9983 L · kg-1和212~2657 L · kg-1.但是,与其他卤代有机物如有机氯农药相比,PFOS和PFOA的Kd值要小几个数量级,说明PFOS和PFOA比其他卤代有机物不易分配于污泥中(Arvaniti et al., 2012).
| 表3 PFOS和PFOA在污水和污泥中的分配系数 Table 3 Distribution coefficient values,Kd for PFOS and PFOA in wastewater and sludge |
从表 3可以看出,PFOA在不同处理阶段的Kd值差别不大,但PFOS在不同处理阶段Kd值差别较大,并且在好氧池最高,其次为缺氧池和二沉池,在厌氧池最低.文献报道表明,分配系数Kd变化范围较大的原因,与污泥性质、污水中污染物的浓度和不同污泥停留时间等因素相关(Arvaniti et al., 2012; Zhou et al., 2010).而Yu等(2009)的报道则称,不同类型的污泥对PFOS和PFOA的分配系数没有太大影响.Arvaniti等(2012)报道了PFCs在初沉池、好氧池和二沉池中的泥水分配系数,结果也显示,对3种污泥而言PFOA的Kd值差别不大,但PFOS在好氧池中的Kd值较高.Arvaniti等(2012)又对此现象做出了推测,不同的污泥性质如pH和所含离子浓度等会对Kd值造成影响.Higgins等(2006)对PFOS在河水和沉积物之间的分配进行了研究,发现除了pH和盐度等因素外,沉积物的有机碳含量也是关键因素之一.好氧池水中的溶解氧含量较高,污水的处理率较高,故水中的有机物含量较低,被污泥吸附待降解的有机物含量较高,所以PFOS更易分布于污泥相中.此外,表 3中的数据也显示,PFOS的Kd值较PFOA高,这说明与PFOA相比,更多的PFOS会吸附于污泥中.
3.3 PFOS和PFOA的质量流分析各构筑物PFOS和PFOA质量流的计算方法如下所示:
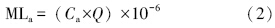
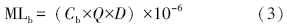
S1~S6和S8夏季流量为9000 m3 · d-1,冬季为70000 m3 · d-1,S7为二沉池回流量及剩余污泥量,夏季流量为12000 m3 · d-1,冬季流量为11700 m3 · d-1.卢沟桥污水处理厂各工艺流程中PFOS和PFOA的质量流如图 3所示,计算误差在16%之内.进水中的PFOS和PFOA部分吸附于SS中,夏季进水中PFOS和PFOA的总负荷分别为12.3 g · d-1和2.4 g · d-1,出水中PFOS和PFOA的总负荷分别为7.0 g · d-1和3.1 g · d-1.除了出水以外,二沉池污泥的约10%污泥作为剩余污泥排出,PFOS和PFOA在二沉池负荷的10%分别为1.2 g · d-1和0.2 g · d-1,也排出到环境中,所以,夏季PFOS和PFOA进入环境中的总负荷量分别为8.2 g · d-1和3.3 g · d-1.同样的,冬季PFOS和PFOA在进水中的负荷分别为12.7 g · d-1和5.5 g · d-1,通过污水处理厂进入环境中的总负荷量(包括出水和剩余污泥)分别为12.9 g · d-1和6.6 g · d-1.可见,进入污水处理厂的PFOS和PFOA的负荷在冬夏季的差别不大,但通过污水厂进入环境中的负荷冬季略高于夏季.无论是在夏季还是冬季,PFOS和PFOA的质量流从进入污水处理厂到排出的过程中并没有明显的减少,反而出现了上升的现象,结果与文献报道相符(Schultz et al., 2006;Becker et al., 2008).
 |
| 图 3 夏季和冬季PFOS和PFOA在污水处理的各工艺阶段的质量流变化 Fig. 3 Mass flow of PFOS and PFOA in wastewater and sludge during each treatment process in summer and winter |
经过初沉池后,污染物吸附于污泥中,水相的质量负荷有所下降,夏季水相和固相中PFOS和PFOA的总负荷在污水处理过程中的变化均不明显.夏季进行污水生物处理的过程中,污泥中PFOS的负荷占泥水混合物总负荷的70%以上,其中,在二沉池污泥(S7)中占到了90%的负荷量;PFOA也有类似的特点,污泥中的负荷占泥水混合物总负荷的50%以上,并且在二沉池污泥(S7)中比例达到最高,为82%.在Loganathan等(2007)对肯塔基的一座污水处理厂的研究中,春、夏和冬季经污水处理厂污泥排入环境中的PFOS的污染负荷占到总负荷的90%以上.PFOA在泥水混合物中的总负荷在生物处理的各阶段变化不大,夏季和冬季的情况类似,而PFOS在夏季和冬季的情况较为不同.在夏季,PFOS在污水处理各阶段的质量负荷呈下降趋势,进入初沉池后即达到最大,随后一直降低.而在冬季,从进水到好氧池2,PFOS的质量负荷呈增加的趋势,在好氧池2达到最大.这可能是因为由于污水中存在可以降解为PFOS的前驱物质,冬季污泥停留时间较长,降解产生的PFOS在污泥中进行积累从而产生质量负荷逐渐增大的现象.另外,采样的方式有可能也是影响质量流分析的重要因素之一(Yu et al., 2009).文献研究认为,在抓取的采样方式下,质量流发生30%以上的变化才能表示发生了明显的变化(Loganathan et al., 2007;Yu et al., 2009).本研究所采取的抓取采样的方式,有可能在浓度较大或者较小的时候进行的采样,所以有可能增加浓度的变化范围.质量流的分析是根据浓度计算的,所以相应的质量流的变化范围也有可能增大.
4 结论(Conclusions)1)通过对PFOS和PFOA在倒置A2O工艺各工艺段污水和污泥中的浓度检测,以及对泥水分配的分析和对质量流的计算,结果发现,卢沟桥污水处理厂污水和污泥中PFOS的浓度大于PFOA,这与进水的来源主要为市政污水有关.
2)整个工艺流程污水和污泥中PFOA的浓度及质量流均基本保持不变,受泥水分配的影响,PFOS的浓度和质量流在工艺流程的各阶段变化较大.
3)PFOS的泥水分配系数Kd较PFOA高,说明PFOS较PFOA更易分布于污泥中,并且PFOS的Kd值受污泥种类和特性的影响较大.对质量流的分析也表明,PFOS主要分布于泥相中.
4)PFOA的浓度及质量流在夏季和冬季之间差别不大,但PFOS的变化较为明显,可能是因为冬夏季污泥特性有所不同,并且冬季比夏季污泥停留时间长,PFOS在污泥中积累所致.
PFOS和PFOA经过污水处理厂后,以出水和剩余污泥的形式进入环境中.污水处理厂作为全氟化合物的污染源之一,出水中的PFOS和PFOA已经引起了关注.根据质量流的分析可以看出,更多的污染物是随剩余污泥排出,如果污泥处理处置措施不合适,则会导致PFOS和PFOA进入环境水体和土壤中,对生物和人体造成潜在的危害,所以,污泥处理处置过程中PFOS和PFOA的迁移转化行为值得进一步进行深入研究.
| [1] | Ahrens L, Shoeib M, Harner T, et al. 2011. Wastewater treatment plant and landfills as sources of polyfluoroalkyl compounds to the atmosphere[J]. Environmental Science & Technology, 45(19): 8098-8105 |
| [2] | Arvaniti O S, Ventouri E I, Stasinakis A S, et al. 2012. Occurrence of different classes of perfluorinated compounds in Greek wastewater treatment plants and determination of their solid-water distribution coefficients[J]. Journal of Hazardous Materials, 239-240: 24-31 |
| [3] | Becker A M, Gerstmann S, Frank H. 2008. Perfluorooctane surfactants in waste waters, the major source of river pollution[J]. Chemosphere, 72(1): 115-121 |
| [4] | Bossi R, Strand J, Sortkjr O, et al. 2008. Perfluoroalkyl compounds in Danish wastewater treatment plants and aquatic environments[J]. Environment International, 34(4): 443-450 |
| [5] | Boulanger B, Vargo J D, Schnoor J L, et al. 2005. Evaluation of perfluorooctane surfactants in a wastewater treatment system and in a commercial surface protection product[J]. Environmental Science & Technology, 39(15): 5524-5530 |
| [6] | BSI (British Standards Institution). 2009. BS ISO 25101: 2009 Water quality-Determination of perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA) - Method for unfiltered samples using solid phase extraction and liquid chromatography/mass spectrometry[S]. London, UK: BSI |
| [7] | Busch J, Ahrens L, Sturm R, et al. 2010. Polyfluoroalkyl compounds in landfill leachates[J]. Environmental Pollution, 158(5): 1467-1471 |
| [8] | Butenhoff J L, Chang S C, Olsen G W, et al. 2012. Chronic dietary toxicity and carcinogenicity study with potassium perfluorooctanesulfonate in Sprague Dawley rats[J]. Toxicology, 293(1/3): 1-15 |
| [9] | Calafat A M, Kuklenyik Z, Caudill S P, et al. 2006a. Perfluorochemicals in pooled serum samples from United States residents in 2001 and 2002[J]. Environmental Science & Technology, 40(7): 2128-2134 |
| [10] | Calafat A M, Needham L L, Kuklenyik Z, et al. 2006b. Perfluorinated chemicals in selected residents of the American continent[J]. Chemosphere, 63(3): 490-496 |
| [11] | Chen H, Zhang C, Han J B, et al. 2012. PFOS and PFOA in influents, effluents, and biosolids of Chinese wastewater treatment plants and effluent-receiving marine environments[J]. Environmental Pollution, 170: 26-31 |
| [12] | Guo R, Sim W, Lee E S, et al. 2010. Evaluation of the fate of perfluoroalkyl compounds in wastewater treatment plants[J]. Water Research, 44(11): 3476-3486 |
| [13] | Higgins C P, Field J A, Criddle C S, et al. 2005. Quantitative determination of perfluorochemicals in sediments and domestic sludge[J]. Environmental Science & Technology, 39(11): 3946-3956 |
| [14] | Higgins C P, Luthy R G. 2006. Sorption of perfluorinated surfactants on sediments[J]. Environmental Science & Technology, 40(23): 7251-7256 |
| [15] | Huset C A, Chiaia A C, Barofsky D F, et al. 2008. Occurrence and mass flows of fluorochemicals in the Glatt valley watershed, Switzerland[J]. Environmental Science & Technology, 42(17): 6369-6377 |
| [16] | Jensen A A, Leffers H. 2008. Emerging endocrine disrupters: perfluoroalkylated substances[J]. International Journal of Andrology, 31(2): 161-169 |
| [17] | Kannan K, Corsolini S, Falandysz J, et al. 2004. Perfluorooctanesulfonate and related fluorochemicals in human blood from several countries[J]. Environmental Science & Technology, 38(17): 4489-4495 |
| [18] | Kim S, Im J, Kang Y, et al. 2012. Wastewater treatment plants (WWTPs)-derived national discharge loads of perfluorinated compounds (PFCs)[J]. Journal of Hazardous Materials, 201-202: 82-91 |
| [19] | Kunacheva C, Tanaka S, Fujii S, et al. 2011. Mass flows of perfluorinated compounds (PFCs) in central wastewater treatment plants of industrial zones in Thailand[J]. Chemosphere, 83(6): 737-744 |
| [20] | Lin A Y, Panchangam S C, Ciou P S. 2010. High levels of perfluorochemicals in Taiwan's wastewater treatment plants and downstream rivers pose great risk to local aquatic ecosystems[J]. Chemosphere, 80(10): 1167-1174 |
| [21] | Lin H, Niu J F, Ding S Y, et al. 2012. Electrochemical degradation of perfluorooctanoic acid (PFOA) by Ti/SnO2-Sb, Ti/SnO2-Sb/PbO2 and Ti/SnO2-Sb/MnO2 anodes[J]. Water Research, 46(7): 2281-2289 |
| [22] | Loganathan B G, Sajwan K S, Sinclair E, et al. 2007. Perfluoroalkyl sulfonates and perfluorocarboxylates in two wastewater treatment facilities in Kentucky and Georgia[J]. Water Research, 41(20): 4611-4620 |
| [23] | Murakami M, Shinohara H, Takada H. 2009. Evaluation of wastewater and street runoff as sources of perfluorinated surfactants (PFSs)[J]. Chemosphere, 74(4): 487-493 |
| [24] | OECD (Organization for Economic Co-operation and Development). 2002. Co-operation on Existing Chemicals Hazard Assessment of Perfluorooctane Sulfonate (PFOS) and its salts[R]. ENV/JM/RD(2002)17/FINAL. Paris, France: OECD. 12-14 |
| [25] | Schultz M M, Barofsky D F, Field J A. 2006a. Quantitative determination of fluorinated alkyl substances by large-volume-injection liquid chromatography tandem mass spectrometry characterization of municipal wastewaters[J]. Environmental Science & Technology, 40(1): 289-295 |
| [26] | Schultz M M, Higgins C P, Huset C A, et al. 2006b. Fluorochemical mass flows in a municipal wastewater treatment facility[J]. Environmental Science & Technology, 40(23): 7350-7357 |
| [27] | Sinclair E, Kannan K. 2006. Mass loading and fate of perfluoroalkyl surfactants in wastewater treatment plants[J]. Environmental Science & Technology, 40(5): 1408-1414 |
| [28] | Skutlarek D, Exner M, Farber H. 2006. Perfluorinated surfactants in surface and drinking water[J]. Environmental Science and Pollution Research, 13(5): 299-307 |
| [29] | Sun H W, Li F S, Zhang T, et al. 2011. Perfluorinated compounds in surface waters and WWTPs in Shenyang, China: Mass flows and source analysis[J]. Water Research, 45(15): 4483-4490 |
| [30] | Takazawa Y, Nishino T, Sasaki Y, et al. 2009. Occurrence and distribution of perfluorooctane sulfonate and perfluorooctanoic acid in the rivers of Tokyo[J]. Water, Air, & Soil Pollution, 202(1): 57-67 |
| [31] | Wang N, Szostek B, Folsom P W, et al. 2005. Aerobic biotransformation of 14C-labeled 8-2 telomer B alcohol by activated sludge from a domestic sewage treatment plant[J]. Environmental Science & Technology, 39(2): 531-538 |
| [32] | Yan H, Zhang C J, Zhou Q, et al. 2012. Short-and long-chain perfluorinated acids in sewage sludge from Shanghai, China[J]. Chemosphere, 88(11): 1300-1305 |
| [33] | Yu J, Hu J, Tanaka S, et al. 2009. Perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA) in sewage treatment plants[J]. Water Research, 43(9): 2399-2408 |
| [34] | Zhou Q, Deng S B, Zhang Q Y, et al. 2010. Sorption of perfluorooctane sulfonate and perfluorooctanoate on activated sludge[J]. Chemosphere, 81(4): 453-458 |