 2014, Vol. 34
2014, Vol. 34



煤炭是我国的主要能源之一,其燃烧将排放出大量的氮氧化物(NOx)气体,而氮氧化物的存在则会造成酸雨、光化学烟雾及臭氧层空洞等危害(Zhu et al., 2009; Bowker et al., 2008),因此,研究适合我国工厂烟气排放系统特点的烟气脱硝技术,对我国大气污染的治理有着举足轻重的意义.目前,工业烟气尤其是燃煤烟气中氮氧化物的脱除技术主要以选择性催化还原(SCR)技术为代表(Qi et al., 2004),主要通过向烟气排放系统中喷入NH3并在SCR催化剂的作用下将氮氧化物还原为N2.尽管该方法在工业中的应用较为成熟,但会消耗大量工业NH3,操作运行费用较大,还原产物N2也没有回收利用的价值,且在处理过程中需严格控制NH3的化学计量比,以避免氨逃逸造成二次污染.此外,为避免烟气中SO2的毒化,SCR技术适用的温度窗口为350 ℃以上,故在中低温区的使用受到较大的技术限制.因此,作为新型的脱硝技术——氮氧化物的选择性催化氧化技术(SCO)应运而生.
NO是烟气中NOx的主要成分,NO除生成络合物外,无论在水中或碱液中都不易被吸收,而NO2在水中的溶解度很高,可选择合适的碱或盐溶液(如Na2SO4)进行吸收(Luo et al., 2005).因此,采用催化技术将气相中的NO氧化为NO2然后利用碱液进行吸收脱除是一条较为可行的技术路线,并且NO2能够与另一污染性气体SO2在同一装置内吸收脱除,吸收产生的化工产品可以回收利用,综合经济效益将显著提高.故该技术将是极具竞争力的烟气同时脱硫脱硝技术.氮氧化物的氧化-吸收法不会造成二次污染,且有望实现湿法或者半干法脱硫脱硝一体化.因此,研制出高效低温氧化脱硝催化剂对于我国燃煤烟气净化技术的发展具有重要意义.
研究表明,采用Pt作为活性组分的催化剂具有较高的低温氧化脱硝效率(Després et al., 2004; Schmitz et al., 2006; Olsson et al., 2002),但Pt价格昂贵且易于失活,因此,找到价廉高效的替代品成为当今研究的热点.过渡金属具有较好的氧化还原能力,可用来替代Pt,其中,锰基催化剂在SCR反应中表现出较高活性而被较多用于催化氧化脱硝.Li 等(2012)采用浸渍法制备MnOx/TiO2催化剂,在反应温度为250 ℃时,NO氧化率达到48%.莫建红等(2007)采用共沉淀-浸渍法制备了Mn/Co-Ba-Al-O催化剂,在反应温度为225 ℃左右时,NOx的氧化率(NO2/NOx)达到50%~60%.Zhang等(2008)采用共沉淀法制备Mn-Fe/MPS催化剂,在280 ℃时,NO转化率达到70%.
氧化锆是一种同时拥有酸碱性、氧化性和还原性的过渡金属氧化物,且为P-型半导体材料,作为催化剂载体时易于与活性组分产生相互作用,提高催化剂的活性(杨淑梅等,2003).氧化锆与其他常见载体(TiO2、SiO2、Al2O3)相比表现出较好的活性(曲玲玲等,2009).但以氧化锆作为载体制备催化剂并用于催化氧化NO的研究并不多见,因此,本文采用氧化锆作为载体负载锰氧化物制备一种用于SCO反应的新型Mn/ZrO2催化剂,对其催化氧化NO性能进行一系列活性评价,并通过表征分析表明了NO的氧化反应机理.
2 实验部分(Experimental section) 2.1 实验材料与试剂主要试剂:50%硝酸锰溶液(Mn(NO3)2);醋酸锰(Mn(CH3COO)2 · 4H2O);氧化锆(ZrO2)、聚乙二醇(PEG-6000)、氨水均为分析纯,由国药集团化学试剂有限公司提供.实验所用气体均由南京特种气体有限公司提供.
2.2 催化剂活性评价催化剂的脱硝活性评价在常压固定床反应器中进行,反应器采用内径为15 mm、高为80 mm的竖式石英管,由管式电炉加热.活性评价装置由模拟烟气、固定床反应器和分析检测三大部分构成,反应流程如图 1所示.催化剂用量为2 g,模拟烟气由NO、O2、Ar混合而成,反应温度为200~400 ℃.采用德国TESTO公司生产的testo340型烟气分析仪分别检测反应器进出口处NO、NO2的浓度.
 |
| 图 1 脱硝活性评价装置 Fig. 1 Schematic diagram of experimental apparatus for activity test |
催化剂脱硝活性以NO转化率(XNO)和NO2选择性(SNO2)衡量,计算公式分别为:
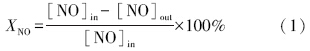
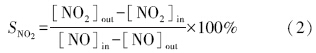
等体积浸渍法:向氧化锆粉末(AR)中加入1%(质量分数)田菁粉,充分搅拌均匀后加入适量蒸馏水,搅拌形成粘性浆料,置于60 ℃烘箱中干燥得到块状固体,将其粉碎筛分,得到粒径为20~40目的颗粒,作为催化剂载体.然后将一定量的催化剂载体分别浸渍到不同浓度的硝酸锰或醋酸锰溶液中,静置24 h,然后在60 ℃干燥3 h,110 ℃干燥12 h,最后置于空气中于450 ℃煅烧3 h,制得Mnx(-MA)/ZrO2/450-IM催化剂(其中,x代表催化剂上Mn元素占载体的质量分数,MA表示醋酸锰作为前驱体(无标注的表示硝酸锰作为前驱体),450表示煅烧温度,IM表示等体积浸渍法).
共沉淀法:将ZrOCl2 · 8H2O溶液溶于水中,配制成 1 mol · L-1的溶液,加入适量的PEG-6000分散剂,在45 ℃恒温搅拌下用1 mol · L-1氨水缓慢中和至pH=8.0,恒温搅拌30 min,过滤、洗涤,将所得沉淀物在80 ℃烘干保温2 h,然后在空气中于400 ℃焙烧2 h,粉碎过筛,得到20~40目颗粒作为载体备用.然后取所需量的Mn(NO3)2溶液加入氨水调节pH至10,加入上述载体维持pH不变,70 ℃恒温搅拌2 h,过滤、洗涤,将所得沉淀物在80 ℃烘干保温2 h,再在空气中于450 ℃焙烧3 h,制得催化剂Mnx/ZrO2/450-CP催化剂(其中,x代表催化剂上Mn元素占载体的质量分数,450表示煅烧温度,CP表示共沉淀法).
2.4 催化剂的表征 2.4.1 BET比表面积测定催化剂的BET比表面积采用全自动比表面积仪测定N2吸附等温线,经BET方程计算得到.液氮为吸附介质,吸附温度77 K,测试前所有样品均在不同的温度下干燥预处理.比表面积通过Brunaner Emmett Teller(BET)方程计算得到.
2.4.2 扫面电镜(SEM)SEM在中国科技大学理化测试中心采用FEI公司Sirion200扫描仪完成,最大分辨率1.5 nm(>10 kV),加速电压0.2~30 kV;放大倍数74~300000.
2.4.3 X射线衍射(XRD)催化剂的X射线衍射(XRD)表征用日本理学D/max-rB型X射线衍射仪完成,Cu Kα射线,管电压40 kV,管电流100 mA,扫描速度4° · min-1,扫描范围2θ=3°~80°.
2.4.4 X射线光电子能谱(XPS)催化剂的X射线光电子能谱(XPS)表征采用美国Therm公司ESCALAB250型X射线光电子能谱仪完成,Al Kα射线,管电压1486.6 eV,测定催化剂表面原子状态.
2.4.5 程序升温脱附(TPD)程序升温脱附(TPD)实验在石英微反应管中常压进行.取2.0 g样品放入Φ15 mm石英管中,通入100 mL · min-1Ar气吹扫1 h,然后在50 ℃温度下切换成反应气NO,用Ar气作平衡气,总流量为350 mL · min-1,待吸附饱和后用Ar气吹扫以除去表面可逆吸附或弱吸附组分.然后在Ar气氛围中以5 ℃ · min-1速率从50 ℃程序升温至550 ℃,反应过程中反应器出口气体通过testo 350烟气分析仪进行检测.
2.4.6 程序升温表面反应(TPSR)催化剂的TPSR实验在石英微反应管中常压进行.取2.0 g样品放入Φ15mm石英管中,通入100 mL · min-1 Ar气吹扫1 h,然后在50 ℃温度下切换成反应气NO,用Ar气作平衡气,总流量为350 mL · min-1,待吸附饱和后用Ar气吹以扫除去表面可逆吸附或弱吸附组分.然后切断NO通入10%O2,Ar气为平衡气,总流量保持不变,以5 ℃ · min-1的速率程序升温至550 ℃,反应过程中反应器出口气体通过testo 350烟气分析仪进行检测.
3 实验结果与讨论(Results and discussion) 3.1 制备条件对催化剂活性的影响 3.1.1 负载方法及前驱体对催化剂活性的影响等体积浸渍法和共沉淀法是制备催化剂较为常用的两种方法,本文考察了两种制备方法对催化剂活性的影响,结果如图 2所示.从图中可以观察到,采用等体积浸渍法制备的催化剂其活性优于以共沉淀法制备的催化剂,尤其是在220~350 ℃的温度区间内.在250 ℃反应温度下,Mn8/ZrO2/450-IM催化剂对NO转化率达到63%,而Mn8/ZrO2/450-CP催化剂对NO转化率只有45%.同时共沉淀法在制备工艺上相对较为复杂,因此,在后面的催化剂制备中选择采用等体积浸渍法.
 |
| 图 2 不同制备方法对Mn8/ZrO2/450催化剂活性的影响(反应条件:0.06% NO,10% O2,Ar作平衡气,空速为15000h-1) Fig. 2 NO conversion over Mn8/ZrO2/450 prepared by different methods(react condition: 0.06% NO,10% O2,balance Ar,GHSV=15000 h-1) |
图 3为不同前驱体对Mn8/ZrO2/450-IM催化剂活性的影响.从图中可以看出,在所考察反应温度范围内,以硝酸锰作为前躯体制备的催化剂活性明显优于以醋酸锰作为前躯体制备的催化剂.在反应温度为300 ℃时,Mn8/ZrO2/450-IM催化剂的NO转化率可达到84%,但Mn8-MA/ZrO2/450-IM催化剂的NO转化率只有60%.这一结果说明锰基催化剂采用不同前驱体制备的催化剂可能形成不同的表面活性组分形态,从而影响催化剂的活性.Eunseuk等(2012)研究发现,采用硝酸锰作为前驱体制备的催化剂经煅烧后主要形成MnO2,而采用醋酸锰作为前驱体时主要形成的为Mn2O3.
 |
| 图 3 不同前驱体对Mn8/ZrO2/450-IM催化剂活性的影响(反应条件:0.06% NO,10% O2,Ar作平衡气,空速为15000 h-1) Fig. 3 NO conversion over Mn8/ZrO2/450-IM with different precursors(react condition: 0.06% NO,10% O2,balance Ar,GHSV=15000 h-1) |
图 4为不同Mn负载量对催化剂活性的影响.从图中可以看出,随锰负载量的增加(由2%增加至6%),催化剂活性逐渐增加,当锰负载量增加至8%后继续增加锰氧化物负载量时,NO转化率变化不大,说明催化剂具有较好的稳定性.活性组分负载量对催化剂活性有着至关重要的作用,活性组分负载量过少则在载体表面不能形成有效的活性位,但过多则可能会覆盖活性位或造成孔道堵塞,也有可能出现表面体相,分散度降低.同时由图 4可以看出,催化剂具有较宽的温度窗口,在250~350 ℃反应温度范围内均表现出较佳的脱硝活性,且均在300 ℃反应温度下脱硝活性达到最佳.同时,为了表明实验数据的精密度,对Mn8/ZrO2/450-IM催化剂对NO的转化率曲线进行误差线的标注,结果如图 5所示.由图 5可以得出,实验数据精密度良好.为进一步表明不同负载量催化剂在最佳反应温度下的脱硝活性,将不同负载量催化剂在300 ℃反应温度下对NO转化率及NO2选择性示于图 6.由图 6a可以发现,催化剂活性较为稳定,不同负载量催化剂在300 ℃反应温度下对NO的转化率基本稳定在80%左右,同时发现锰负载量为8%的催化剂在300 ℃反应温度下具有最高的NO转化率,可达84%.同时,由图 6b可以发现,不同负载量催化剂在300 ℃反应温度下对NO2的选择性可达到90%以上(除Mn4/ZrO2/450-IM催化剂对NO2的选择性为84%),因此,可以认为NO催化氧化反应的主要产物为NO2,其他副反应产物可以不予考虑.为方便研究,选择锰负载量为8%的催化剂进行下一步的研究.
 |
| 图 4 锰负载量对NO转化率的影响(反应条件:0.06% NO,10% O2,Ar作平衡气,空速为15000 h-1) Fig. 4 The NO removal activity of catalysts with different loadings of active component(react condition: 0.06% NO,10% O2,balance Ar,GHSV=15000 h-1) |
 |
| 图 5 Mn8/ZrO2/450-IM催化剂对NO转化率的误差分析(反应条件:0.06% NO,10% O2,Ar作平衡气,空速为15000 h-1) Fig. 5 Error analysis of NO conversion over Mn8/ZrO2/450-IM catalysts(react condition: 0.06% NO,10% O2,balance Ar,GHSV=15000 h-1) |
 |
| 图 6 300 ℃反应温度下不同负载量对Mnx/ZrO2/450-IM催化剂脱硝活性的影响(反应条件:0.06% NO,10% O2,Ar作平衡气,空速为15000 h-1) Fig. 6 The NO removal activity of catalysts with different loadings of active component at 300 ℃(react condition: 0.06% NO,10% O2,balance Ar,GHSV=15000 h-1) |
反应温度是NO催化氧化的重要影响因素,是催化氧化反应的关键.NO氧化反应为放热反应,随反应温度的升高平衡转化率逐渐下降.由图 4可得,随着反应温度的升高NO转化率逐渐增大,反应温度达到300 ℃时,NO转化率曲线达到最高点,继续提高反应温度,NO氧化率逐渐下降.可能的原因是低温时NO氧化反应的产物NO2在催化剂表面上呈现强吸附,对反应有阻碍作用;而温度升高后,NO2逐渐脱附,催化剂表面NO2浓度降低,NO氧化反应得以顺利进行,反应速率加快;至300 ℃左右NO2脱附速率达到最大,NO的转化率也达到最高(在后面的分析中得到验证).Mnx/ZrO2/450-IM催化剂的最佳反应温度为300 ℃,因此,我们选择300 ℃反应温度进行后续研究.
3.2.2 空速对催化剂活性的影响NO的催化氧化过程必须选择适合的空速.空速过大时,虽然催化剂的生产能力有所增加,但因气固接触时间减少,不利于反应气体在催化剂空隙中的扩散、吸附和产物的解吸、扩散,导致反应不完全,NO转化率下降.空速较小时,反应物可与催化剂充分接触,反应较完全,转化率较高,但空速过低将造成单位体积催化剂处理量下降,也会导致实际应用成本上升.因此,空速大小对SCO脱硝技术有着重要的影响.本文研究了空速对Mn8/ZrO2/450-IM催化剂脱SCO活性的影响,结果如图 7所示.由图可以看出,空速由5000 h-1增加到18000 h-1时,催化剂活性保持稳定,在空速增加到24000 h-1时,催化剂活性仅稍微有所下降,由80%下降到了76%.这表明本研究中制备的催化剂具有较高的稳定性.
 |
| 图 7 空速对Mn8/ZrO2/450-IM催化剂脱硝活性的影响(反应条件:0.06% NO,10% O2,Ar作平衡气,总流量为350 mL · min-1) Fig. 7 Effect of GHSV on NO conversion over Mn8/ZrO2/450-IM(reaction condition: 0.06% NO,10% O2,balance Ar,gas flow rate of 350 mL · min-1) |
实际生产中根据不同的燃烧状况,锅炉烟气中会混有一定量的空气,空气量的多少对NO的催化氧化反应有着一定的影响,因此,本文考察了不同氧含量对催化剂活性的影响.如图 8所示,不通氧气时,NO转化率很低,通入3%(体积分数)氧气时,NO转化率达到73%,继续增大氧气量至5%时,NO转化率接近80%,继续增加O2通入量,NO转化率基本保持稳定.可以认为O2量达到5%时,O2对NO催化氧化动力学反应为零级.根据Iwamoto等(1978)的研究,O2在P-型过渡金属氧化物表面存在强吸附,呈现多种吸附物种,主要以O-形式存在.因此,O2浓度的增加能增大催化剂表面的氧原子含量,从而可以提高氧化转化率,但当氧浓度增加到一定值时,催化剂表面氧的吸附达到饱和,氧化效率趋于稳定.通常,在燃煤锅炉中,烟气中氧含量为8%~10%(郑足红等,2008),因此,本实验所制备的催化剂适合应用于锅炉烟气中NO的脱除.
 |
| 图 8 O2含量对Mn8/ZrO2/450-IM催化剂活性的影响(反应条件:0.06% NO,Ar作平衡气,空速14862 h-1,总流量为350 mL · min-1) Fig. 8 Effect of O2 concentration on NO conversion over Mn8/ZrO2/450-IM(react condition: 0.06% NO,balance Ar,GHSV=14862 h-1,gas flow rate of 350 mL · min-1) |
催化剂比表面积的大小对催化剂活性有着一定的影响,大的比表面积更有利于活性组分的负载和反应气体的吸附.本文对不同负载量催化剂进行了BET测定(表 1).由表 1可以看出,催化剂比表面积都比较小,锰负载量的变化对催化剂比表面积的影响并不显著,且比表面积的细微变化没有明显的规律性.与上文所述催化剂活性的变化规律相比较,比表面积的变化与催化活性间没有直接的关联性,说明该催化剂的催化氧化活性对比表面积大小并不敏感,比表面积的大小并非影响催化剂活性的直接因素.
| 表1 Mnx/ZrO2/450-IM催化剂的比表面积 Table 1 BET surface area of Mnx/ZrO2/450-IM catalysts |
图 9给出了分别采用等体积浸渍法和共沉淀法制备的Mn8/ZrO2/450催化剂的SEM图.由图 9a可知,采用等体积浸渍法制备的催化剂表面整体由小块状结构组成,表面多孔,一层纳米级小颗粒松散均匀分布在催化剂表面,它们应该为锰氧化物.由图 9b可知,采用共沉淀法制备的催化剂表面整体呈大块状结构,表面较为平坦少孔,纳米级颗粒较为密集地堆积在催化剂表面.以上结果表明,采用不同制备方法制备的催化剂表面形貌差异较大,氧化物分散程度也不同,这可能是造成催化剂SCO性能差异的原因之一.采用等体积浸渍法制备的催化剂表面颗粒分散均匀,载体表面疏松多孔,这可能与其有较好的活性有关.
 |
| 图 9 不同制备方法制备催化剂的SEM图(a.Mn8 /ZrO2/450-IM,b. Mn8 /ZrO2/450-CP) Fig. 9 SEM morphology of Mn8 /ZrO2/450 catalysts prepared by different methods(a.Mn8 /ZrO2/450-IM,b. Mn8 /ZrO2/450-CP) |
为探究在不同锰氧化物负载量下催化剂表面锰氧化物的负载形态及分散情况,对不同负载量催化剂进行了XRD表征.图 9为不同负载量催化剂的XRD图谱.由氧化锆载体的XRD谱图中,观察到2θ=24.059°、28.180°、31.461°、34.177°、49.278°、50.102°等处的衍射峰,对应的为单斜晶相ZrO2(JCPDS 83-0943).质量分数低于12%的锰负载在氧化锆载体上经450 ℃煅烧后,ZrO2载体的晶相没有发生明显变化,没有检测到锰氧化物的衍射峰;当锰负载量高于12%时单斜晶氧化锆的峰位没有发生变化,但在2θ=37.091°、42.595°处出现了新的微弱的MnO2(JCPDF 14-0644)衍射峰.可以说明,在锰负载量低于12%时,活性组分在催化剂表面处于高度分散状态(或呈无定形态);当锰氧化物负载量增加至12%时,逐渐出现微弱的MnO2晶体,推断出催化剂表面的活性物质主要为MnO2,同时可能会含有高度分散的Mn2O3,这与后面的XPS结果相一致.
3.6 XPS分析为进一步探讨催化剂表面锰元素和氧元素的存在形态,对Mn8/ZrO2/450-IM催化剂进行了XPS表征.图 10为Mn8/ZrO2/450-IM催化剂表面Mn 2p3/2和O 1s的XPS谱图.图 10a为锰元素的XPS谱图,锰元素主要以Mn3+和Mn4+形式存在,Mn2O3的结合能为 641.3~641.7 eV,MnO2的结合能为642.2~643 eV(Li et al., 2012;Wu et al., 2010).图 10b为氧元素的XPS谱图,结合能为529~530 eV对应的为晶格氧(用Oβ表示),结合能为531~533 eV对应的为吸附氧(用Oα表示)(Li et al., 2012).将XPS图谱进行拟合,分析结果示于表 2.由表 2可知,锰元素多数以Mn4+形式存在,其中,Mn3+/Mn4+为0.64,与XRD结果相符合;氧元素主要以晶格氧形态存在,Oα/Oβ为0.264.根据Atribak等(2010)的研究表明,与Mn4+相比Mn3+能提供更多的活性位,更有利于NO的氧化,因此,可以通过改变催化剂的制备条件(比如,制备催化剂时采用Ar气煅烧)来提高催化剂表面Mn3+的含量,从而提高催化剂的氧化脱硝活性.
 |
| 图 10 不同负载量Mnx/ZrO2/450-IM催化剂XRD谱图 Fig. 10 XRD patterns of Mnx/ZrO2/450-IM catalysts with different Mn loadings |
 |
| 图 11 Mn8/ZrO2/450-IM的XPS谱图 Fig. 11 XPS patterns of Mn8/ZrO2/450-IM catalyst |
| 表2 Mn8/ZrO2/450-IM催化剂表面原子浓度及原子比 Table 2 Surface atomic concentration and surface atom ratio of Mn8/ZrO2 catalyst |
上文对氧化锆负载锰氧化物催化剂的活性评价及表征结果显示,氧化锆锰基催化剂具有较高的NO催化氧化脱除活性,使得烟气NOx气相催化氧化-碱液吸收脱除技术的应用具有较高的可行性.本文采用NO在催化剂表面的吸脱附及暂态反应技术对NO催化氧化反应机理进行研究.图 12为NO的吸脱附曲线和程序升温表面反应(TPSR)曲线.从图 12a可见,NO在催化剂表面的吸附总量较小,在25 min左右达到吸附平衡.由图 12b可以看出,NO在Mn8/ZrO2/450-IM催化剂上主要存在4个热稳定性差别明显的脱附峰.其中,第1个脱附峰位为120 ℃左右的脱附占主导,热稳定性较低,可能与NO物理吸附或弱吸附产生的亚硝酰基或协同亚硝酰胺有关(Tatsuji et al., 1997);第2个脱附峰位于200~300 ℃的较宽区域,该中温脱附峰可能与一种桥键硝酸盐有关(Tatsuji et al., 1997);第3个脱附峰在360 ℃,为高热稳定性脱附峰,可能与NO强吸附产生的硝酸盐类有关(Kijlstra et al., 1997);第4个脱附峰在500 ℃,也为高温脱附峰,可能与NO吸附产生的另一种硝酸盐类有关.图 13为NO在Mn8/ZrO2/450-IM催化剂上的TPSR曲线.从图中可以看出,TPSR过程中反应器出口的NO2释放曲线存在两个较明显的峰位,在此过程中,尾气中没有检测到NO的逸出,表明吸附在催化剂表面的NO在表面反应中发生了转化而不是部分脱附.表 4所列的TPD过程中NO的总脱附量与TPSR过程中NO2的释放量基本相当,也印证了表面吸附的NO在含氧气氛升温过程中主要以氧化转化为主.但由图 12b与图 13显示的峰位置及峰强度可见,NO的脱附与其TPSR过程中的NO2释放曲线并不对应,具体峰位置如表 4所示.TPD过程中NO的脱附以低温峰位为主,而TPSR过程中NO2的释放以中高温为主.上文所述,活性评价的结果显示,NO的氧化率在300oC左右达到最高点,与TPSR过程中NO2的主要释放峰位相一致.
 |
| 图 12 Mn8/ZrO2/450-IM催化剂对NO的吸附曲线(a)和程序升温脱附曲线(b) Fig. 12 Adsorption(a) and TPD(b)profiles of NO over Mn8/ZrO2 catalyst |
 |
| 图 13 吸附NO至饱和Mn8/ZrO2/450-IM催化剂的TPSR曲线 Fig. 13 TPSR curve of saturated Mn8/ZrO2/450-IM catalyst after NO adsorption |
| 表4 NO的吸脱附和TPSR的具体参数 Table 4 Values of NO adsorption and desorption and TPSR |
综上所述,可以推断在低温下吸附在催化剂表面活性位上的NO与催化剂表面晶格氧接触后快速反应生成NO2,且生成的NO2在催化剂表面呈现出强吸附进而形成硝酸盐(Azambre et al., 2008),占据催化剂的表面活性位,从而影响NO的氧化反应速率;然后随着反应温度的升高硝酸盐逐渐分解为NO2,在催化剂表面实现脱附,至300 ℃时实现最大脱附,催化剂表面的活性中心裸露出来,NO得以继续吸附反应,反应速率达到最大.因此,推测NO在Mn8/ZrO2/450-IM催化剂上氧化反应的速率控制步骤为表面NO2的脱附,其反应机理示意图如图 14所示.
 |
| 图 14 NO在Mn8/ZrO2/450-IM催化剂上反应历程图 Fig. 14 Reaction mechanism diagram of NO over Mn8/ZrO2/450-IM catalyst |
具体反应历程可表述为,NO吸附在催化剂表面(σ表示催化剂表面活性位):

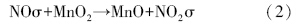

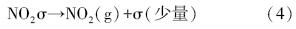
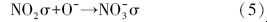

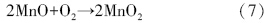
随着温度的升高,催化剂表面形成的硝酸盐逐渐分解生成NO2,在催化剂表面实现脱附,催化剂表面活性中心裸露出来,NO得以继续吸附反应:


通过以上分析及NO的TPD和TPSR曲线,可以得出:在低温下NO的反应速率>NO的吸附速率>NO2的脱附速率,NO2的脱附速率为速率控制步骤,随着反应温度的升高,NO-3逐渐分解,NO2脱附速率加快,NO转化率随之逐渐上升;反应达到300 ℃后,NO2脱附速率>NO吸附速率>NO反应速率,NO反应速率为速率控制步骤,而NO氧化反应为放热反应,随着温度的继续升高,NO氧化速率继续下降,NO转化率也随之下降.分析结果说明:NO以吸附态参与反应,NO2在催化剂表面的脱附速率是由温度控制的,在300 ℃左右时达到最大;Mn8/ZrO2/450-IM催化剂对NO的转化率在低温下主要由NO2的脱附速率控制,在高于300 ℃温度下则主要受NO氧化速率控制.因此,提高负载金属氧化物催化剂脱硝活性的关键是提高低温下NO2在催化剂活性位上的脱附速率,可以考虑采用碱性化合物对催化剂进行改性,转移NO2的吸附位.
4 结论(Conclusions)1)硝酸锰作为前驱体采用等体积浸渍法制备的Mn/ZrO2催化剂具有较高的NO催化氧化活性,在0.06% NO、10% O2、Ar作平衡气、空速为15000 h-1实验条件下,Mn8/ZrO2催化剂在300 ℃最佳反应温度下可达到84%的NO转化率.
2)空速由5000 h-1到24000 h-1之间变化时,Mn8/ZrO2/450-IM催化剂活性保持稳定,NO转化率均为80%左右;O2体积分数达到5%后继续增加氧气通入量,NO转化率保持稳定,为80%左右.
3)对催化剂进行了BET表征,结果表明,催化剂的比表面积与催化剂活性有关但不是影响催化剂活性的直接原因;SEM分析结果表明,采用等体积浸渍法制备的催化剂表面疏松多孔,颗粒分散均匀,这可能与其较好的活性有关;XRD分析表明,锰氧化物在催化剂表面分散良好,且主要的活性物质为MnO2;XPS结果表明,催化剂表面锰元素主要以Mn4+形式存在,氧元素主要以晶格氧形态存在.
4)对催化剂进行了NO吸脱附和TPSR表征,探讨了NO氧化反应机理,结果表明,NO主要以表面吸附态参与反应,在晶格氧的作用下,首先被氧化为中间氧化态物种,进而再分解并以NO2形式脱附.吸附态NO的表面氧化反应为快速步骤,其速率高于NO在反应温度下的脱附速率.NO2的脱附为催化剂上NO的催化氧化过程的决速步骤,低温下NO2难以脱附并占据催化剂活性中心,表现为低温SCR活性较差;高温下NO2的脱附速率加快,宏观表现为NO转化率升高.在高于300 ℃温度下NO转化率逐步下降,可能是由于NO的表面脱附速率逐渐高于表面反应速率,使得部分NO未及氧化便发生表面脱附.
| [1] | Atribak I, Bueno-López A, Carcia Carcia A, et al. 2010. Catalytic activity for soot combustion of birnessite and cryptomelane[J]. Applied Catalysis (B: Environmental), 93: 267-273 |
| [2] | Azambre B, Zenboury L, Delacroix F, et al.2008.Adsorption of NO and NO2 on ceria-zirconia of composition Ce0.69Zr0.31O 2: A DRIFTS study[J]. Catalysis Today, 137: 278-282 |
| [3] | Bowker M. 2008.Automotive catalysis studied by surface science[J]. Chemical Society Reviews, 37(10): 2204-2211 |
| [4] | Després J, Elsener M, Koebel M, et al. 2004. Catalytic oxidation of nitrogen monoxide over Pt/SiO2[J]. Appl Catal B, 50: 73-82 |
| [5] | Eunseuk P, Sungmin C, Juyoung J, et al.2012.Low-temperature NO oxidation over Mn/TiO2 nanocomposite synthesized by chemical vapor condensation: Effects of Mn precursor on the surface Mn species[J]. Microporous and Mesoporous Materials, 163: 96-101 |
| [6] | Kijlstra W S, Brands D S, Poels E K, et al. 1997.Mechanism of the selective catalytic reduction of NO with NH3 over MnOx/Al2O3Ⅰ.Adsorption and desorption of the single reaction components[J].Journal of Catalysis, 171:208-218 |
| [7] | Li X H, Zhang S L, Jia Y, et al. 2012.Selective catalytic oxidation of NO with O2 over Ce-doped MnOx/TiO2 catalysts[J].Journal of Natural Gas Chemistry, 21(1):17-24 |
| [8] | Luo P C, Jiao Z, Wang J F, et al. 2005. NO2 removal from indoor polluted air[J].Chemical Engineering, 33(6):40-43 |
| [9] | Masakazu I, Yukihiro Y, Noboru Y, et al. 1978. Study of metal oxide catalysts by temperature programmed desorption. 4.Oxygen adsorption on various metal oxides[J].The Journal of Physical Chemistry, 82(24):2564-2570 |
| [10] | 莫建红, 童志权, 张俊丰. 2007. Mn/Co-Ba-Al-O催化氧化NO性能研究[J].环境科学学报, 27(11):1793-1798 |
| [11] | Olsson L, Fridell E. 2002.The influence of Pt oxide formation and Pt dispersion on the reactions NO2↔NO+1/2 O2 over Pt/Al2O3 and Pt/BaO/Al2O3[J]. Catal, 210: 340-353 |
| [12] | Qi G S, Yang R T, Chang R.2004.MnOx-CeO2 mixed oxides prepared by co-precipitation for selective catalytic reduction of NO with NH3 at low temperatures[J]. Applied Catalysis (B: Environmental), 51(2):93-106 |
| [13] | 曲玲玲, 周铁桥, 解强, 等. 2009. Ru/ZrO2催化剂高温焙烧对NO催化氧化反应性能的影响[J].环境科学学报, 29(9): 1891-1896 |
| [14] | Schmitz P J, Kudla R J, Drews A R, et al. 2006.NO oxidation over supported Pt: Impact of precursor, support, loading, and processing conditions evaluated via high throughput experi-mentation[J]. Appl Catal B, 67: 246-256 |
| [15] | Tatsuji Y, Albert V.1997.Temperature-programmed desorption of NO adsorbed on Mn2O3 and Mn3O4[J].Applied Catalysis (B:Environmental), 13: 141-155 |
| [16] | Wu Z B, Tang N, Xiao L, et al. 2010. MnOx/TiO2 composite nanoxides synthesized by deposition-precipitation method as a superior catalyst for NO oxidation[J]. Journal of Colloid and Interface Science, 352:143-148 |
| [17] | 杨淑梅, 邓淑华, 黄慧民, 等. 2003. 二氧化锆作载体的催化剂的应用[J].化工时刊, 17(3):1-4 |
| [18] | Zhang J F, Huang Y, Chen X, et al. 2008. Selective catalytic oxidation of NO over iron and manganese oxides supported on mesoporous silica[J]. Journal of Natural Gas Chemistry, 17(3):273-277 |
| [19] | 郑足红, 童华, 童志权, 等.2008.低温高活性NO氧化催化剂Mn-V-Ce/TiO2 的制备与性能[J].过程工程学报, 8(6):1204-1212 |
| [20] | Zhu J J, Thomas A. 2009. Perovskite-type mixed oxides as catalytic material for NO removal[J].Appl Catal B, 92(3/4):225-233 |