 2014, Vol. 34
2014, Vol. 34



2. 环境保护部南京环境科学研究所, 南京 210042
2. Nanjing Institute of Environmental Sciences, Ministry of Environmental Protection, Nanjing 210042
阿特拉津(Atrazine,ATZ)是一种三嗪类除草剂,由于成本低且除草效果好,在世界范围内得到了广泛应用(Sene et al., 2010).ATZ化学性质稳定,在自然条件下降解缓慢,加上多年使用,在土壤中形成了比较严重的残留,并且通过地表径流、淋溶作用进入水体.据报道,美国堪萨斯州地区的井水中ATZ的浓度达7.4 μg · L-1(Buster,1990).王子键等(2002)监测了淮河ATZ的污染状况,发现4个断面的残留量分别为76.4、80.0、72.5、81.3 μg · L-1,远远超过我国国家环保局规定地下水(Ⅰ、Ⅱ类)中ATZ的最大允许浓度3 μg · L-1(GHZBI-1999).叶常明等(2001)监测了白洋淀地区农田表层土壤中ATZ的含量,在3、8和12月份所测得的平均值分别为43.1、64.4、和51.2 ng · L-1.ATZ是潜在的致癌物,也是一种内分泌干扰物,它的存在对水生生态系统和人类饮用水源构成了威胁(Freitas et al., 2008).
基于自由基反应的高级氧化技术是去除有机物的有效手段.常见的高级氧化技术有O3/UV、UV/H2O2、H2O2/O3、芬顿及类芬顿反应等(Barreiro et al., 2007; Chan et al., 2006; Mackul′ak et al., 2011; Tauber et al., 2000; Zhao et al., 2012).它们以产生羟基自由基OH ·(E0=2.8 V)为特点,但对三嗪类物质的降解效果一般(Barreiro et al., 2007).近几年,基于硫酸根自由基(SO· -4)的高级氧化技术由于其自身的一些优势而受到广泛关注(Bennedsen et al., 2012; Ghauch et al., 2012a; Kambhu et al., 2012).和OH ·类似,SO· -4具有较高的氧化电势(E0 = 2.6 V)(Gayathri et al., 2010),但SO· -4比OH ·具更长的寿命(Gayathri et al., 2010),有利于和目标污染物充分接触,并且过硫酸盐不挥发、水溶性好,有利于在土壤和地下水污染处理中的应用.SO· -4由过硫酸盐(S2O2-8)通过加热(Bennedsen et al., 2012; Huang et al., 2002; Liang et al., 2009)、紫外光辐射(Fang et al., 2012; Shih et al., 2012)、过渡金属离子(Niu et al., 2012; Wu et al., 2012)或碱活化(Bennedsen et al., 2012)等方式产生.这几种活化方式各有优缺点.其中,热活化技术能耗较高,但操作简单,在降解机理、产物及限制因素的研究中被大量采用(Ghauch et al., 2012b,杨照荣等,2013),通过热活化研究得到的数据对评价过硫酸盐氧化技术的实际应用具有参考价值.
腐殖酸(HA)是构成土壤、水体中天然有机质的主要成分(Ma et al., 1999).有关HA对过硫酸盐降解污染物的研究还不多.Cl-和CO2-3在水中浓度相对较高,并且研究发现它们可以和SO· -4及OH ·发生反应(Buxton et al., 1999; Liang et al., 2006),与目标污染物竞争氧化剂,从而对去除效率产生影响.因此,本研究利用热活化过硫酸盐技术降解ATZ,着重研究各种因素,包括过硫酸盐浓度、温度、pH、腐殖酸(HA)及常见无机盐离子对水中ATZ降解效率的影响.
2 材料与方法(Materials and methods) 2.1 试剂ATZ购自Sigma-Aldrich公司,过硫酸钾(K2S2O8)、腐殖酸(HA)购自阿拉丁公司,甲醇购自Fisher公司(色谱纯).另外,硫酸、氢氧化钠等所用试剂均为分析纯以上级别.用去离子水配置浓度为 100 μmol · L-1的ATZ、50 mmol · L-1 K2S2O8和100 mg · L-1 的HA母液,保存在冰箱,反应溶液由储备液加去离子水稀释得到.其中,HA储备液的总有机碳(TOC)含量用耶拿multi N/C 3100 TOC仪测定.
2.2 实验方法反应体积为50 mL,ATZ初始浓度为50 μmol · L-1.用H2SO4或NaOH调节溶液pH,置于恒温水浴锅中,加入K2S2O8,并开始计时.每隔20 min,取0.5 mL的样品立即和0.5 mL的100 mmol · L-1 Na2S2O3混合,终止反应,用HPLC分析样品中ATZ的残留.
在以上反应体系中,分别考察S2O2-8浓度(0、0.1、0.5、1.0、2.0 mmol · L-1)、温度(20、30、40、50、60 ℃)、pH(3.0、4.0、5.0、6.0、7.0、8.0、10.0)、HA浓度(2、5、10 mg · L-1)、NaCl浓度(0、5、10、80、200 mmol · L-1)和Na2CO3浓度(0、5、10、50、100 mmol · L-1)对ATZ降解的影响.
自由基鉴定试验中,分别加入甲醇或叔丁醇作为自由基灭活剂,每种试剂取3和30 mmol · L-1两个浓度水平,在同样的条件下,测定ATZ浓度的变化.
2.3 ATZ的定量分析样品中的ATZ用日立(Hitachi)L-2000型高效液相色谱仪(HPLC)进行分析.具体条件为:C18反相色谱柱(Hitachi LaChrom,5 μm × 250 nm × 4.6 mm);流动相为甲醇(70%)和水(30%),流速为1 mL · min-1;柱温为30 ℃;进样量为10 μL;采用二极管阵列检测器(DAD),定量波长为222 nm.
3 结果与讨论(Results and discussion) 3.1 过硫酸盐浓度对降解的影响过硫酸盐的浓度显著影响了ATZ的降解速率,如图 1a所示,ATZ的降解速率随着过硫酸盐浓度的升高而加快.过硫酸盐浓度为0.1 mmol · L-1时,在2 h内ATZ基本没有去除;当过硫酸盐浓度增加到2.0 mmol · L-1,反应2 h后,ATZ的去除率超过98%.此外,ATZ的降解曲线可以用一级反应动力学方程拟合:
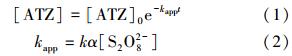
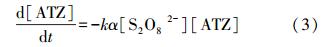
 |
| 图 1 过硫酸盐浓度对过硫酸盐降解ATZ的影响(50 ℃,pH=7.0,[ATZ]0=50 μmol · L-1) Fig. 1 Influence of persulfate concentration on the removal of ATZ by persulfate(50 ℃,pH=7.0,[ATZ]0=50 μmol · L-1) |
考查了20~60 ℃之间ATZ的降解情况,结果如图 2a所示.在各温度下,反应均呈假一级动力学规律.随着温度的升高,降解显著加快,20 ℃时,ATZ的降解半衰期为17385.1 min,60 ℃时,半衰期仅为22.6 min.Ghauch等(2012b)用热活化过硫酸盐技术降解Ibuprofen时也发现,随着温度从50 ℃升到60 ℃,60 min内Ibuprofen的去除率从12%升高到75%.这一方面是因为温度升高增加了S2O2-8的活化效率,增加了溶液中活性自由基的浓度,即方程(2)中的α值;另一方面,反应速率常数k也随温度升高而变大,这是一般的反应动力学规律.kapp随反应温度T的变化如图 2b所示,两者之间的关系遵循阿累尼乌斯方程:
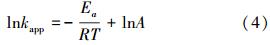
 |
| 图 2 温度对过硫酸盐降解ATZ的影响(pH=7.0,[ATZ]0=50 μmol · L-1,[S2O2-8]0=1.0 mmol · L-1) Fig. 2 Influence of temperature on the removal of ATZ by persulfate(pH=7.0,[ATZ]0=50 μmol · L-1,[S2O2-8]0=1.0 mmol · L-1) |
本研究考察了pH在3.0~10.0范围内过硫酸盐对ATZ的降解情况,结果如图 3所示.pH在3.0~7.0之间时,ATZ在2 h后可达到100%的去除;而在pH=10.0,同样的条件下去除率只有68.6%,显然在碱性条件下ATZ的降解效率低于酸性和中性条件.尽管效率有一定差别,但图 3的结果表明过硫酸盐氧化仍可在广泛的pH条件下起作用,这对于该工艺的实际应用有重要意义.同时,这也是硫酸根自由基氧化对于基于羟基自由基的Fenton氧化的一个显著优势,后者通常要在较低pH(<3.0)下才能获得较高的氧化效率.
 |
| 图 3 pH对过硫酸盐降ATZ的影响(60 ℃,[ATZ]0=50 μmol · L-1,[S2O2-8]0=1.0 mmol · L-1) Fig. 3 Influence of pH on the removal of ATZ by persulfate(60 ℃,[ATZ]0=50 μmol · L-1,[S2O2-8]0=1.0 mmol · L-1) |
碱性条件下ATZ的去除明显慢于酸性和中性条件,很可能由反应体系中自由基的种类和活性在不同pH条件下的差异所导致.在过硫酸盐氧化体系中,可能同时存在SO· -4和OH ·(Huang et al., 2002),前者由过硫酸盐活化直接产生(式(2)),后者可能通过SO· -4二次反应生成(式(3))(Pennington et al., 1968).OH ·对有机物的氧化可通过电子转移、脱氢或加成等机理进行,相对而言,SO· -4对有机物的转化更倾向于通过电子转移途径,因此,SO· -4比OH ·更具选择性(Anipsitakis et al., 2006).
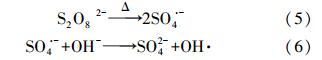
为了确定热活化过硫酸盐体系中自由基的类型,探讨不同pH条件下ATZ降解的机理,本研究分别选择含有R-羟基的乙醇(EtOH)和不含有R-羟基的叔丁醇(TBA)作为自由基清除剂来验证SO· -4和OH ·在溶液中的存在.其中,EtOH为可以同时清除SO· -4和OH ·,而TBA选择性清除OH ·(Anipsitakis et al., 2004).利用两种自由基清除剂对SO· -4和OH ·选择性不同,可检测不同pH条件下溶 液中参与反应的自由基种类.如图 4所示,加入3 mmol · L-1 EtOH,在pH为3.0、7.0和10.0时,对ATZ的降解均有一定的抑制作用,这说明在各pH条件下,ATZ的降解都是由于活性自由基在起作用.当EtOH浓度增加到30 mmol · L-1时,pH为7.0和10.0时,ATZ的降解几乎完全被抑制,但在pH为3.0时,没有完全抑制ATZ的去除,很可能S2O2-8本身也参与了ATZ的氧化.而加入选择性清除OH ·的TBA,浓度为3 mmol · L-1,在pH为3.0和7.0条件下,对ATZ的降解基本没有抑制作用,当TBA浓度增至30 mmol · L-1时,降解略微受到影响.以上现象说明在酸性和中性条件下,溶液中OH ·浓度并不高,对ATZ降解起主导作用的应是SO· -4.但pH为10.0时,TBA对ATZ降解的抑制明显强于在pH为3.0和7.0条件下,这一现象说明碱性条件下溶液中存在OH ·,它对ATZ的降解很可能作了主要的贡献.
 |
| 图 4 在pH为3.0、7.0、10.0时EtOH和TBA作为自由基抑制剂对ATZ降解的影响(60 ℃,[ATZ]0=50 μmol · L-1,[S2O2-8]0=1.0 mmol · L-1) Fig. 4 Effect of EtOH and TBA as radical scavengers(mmol · L-1)on the degradation of ATZ at pH 3.0,7.0,10.0(60 ℃,[ATZ]0=50 μmol · L-1,[S2O2-8]0=1.0 mmol · L-1) |
综上所述,在酸性和中性条件下,溶液中以SO· -4为主,加入选择性抑制OH ·的TBA对反应的影响不大,只有EtOH存在时,才产生明显的自由基灭活作用,对ATZ的去除产生影响.而在碱性条件下,OH ·是参与氧化的主要的活性中间体,无论何种自由基清除剂,均可对其产生有效灭活.溶液中的OH ·可能是OH-在SO· -4氧化作用下生成.相比OH ·,SO· -4具有更长的半衰期(Gayathri et al., 2010),并且过硫酸盐在水相中存在杂质,比如碳酸根离子的情况下,更加稳定(Huang et al., 2002),因此,SO· -4和目标污染物有更多的接触机会.表现在对ATZ的去除上,酸性和中性条件下,效率比碱性溶液中更高.
3.4 HA对降解的影响不同浓度HA对热活化过硫酸盐降解ATZ的影响如图 5所示.在pH为7时,加入HA抑制了ATZ的降解,且随着HA浓度的升高,抑制程度随之增加.未添加HA时,ATZ的降解半衰期为18.6 min,而当加入10 mg · L-1 HA后,ATZ半衰期增大到41.1 min.HA分子中含有羟基、胺基等活性基团,它们和ATZ竞争溶液中的自由基,实际上起了一种自由基清除剂的作用(Ma et al., 1999; Westerhoff et al., 2007).所以,HA对ATZ的去除有负面作用,而且这种影响随HA含量的增加而增加.HA对污染物去除的负面影响在实际污染处理工艺设计中必须充分考虑.
 |
| 图 5 HA对ATZ降解的影响(60 ℃,pH为7.0,[ATZ]0=50 μmol · L-1,[S2O2-8]0=1.0 mmol · L-1) Fig. 5 Effect of HA on the degradation of ATZ(60 ℃,pH 7.0,[ATZ]0=50 μmol · L-1,[S2O2-8]0=1.0 mmol · L-1) |
环境介质中除了广泛存在HA外,还有各种无机阴阳离子,它们对过硫酸盐氧化去除ATZ的影响也是必须研究的重要因素.本研究主要考察了CO2-3和Cl-两种常见的阴离子对ATZ去除的影响.由图 6a可知,当Cl-浓度为10 mmol · L-1甚至更低时,对ATZ的去除起到了明显的促进作用;而当Cl-的浓度升高,则会抑制反应的进行.SO· -4氧化还原电位高达2.6 V,可跟Cl-发生反应,将其氧化生成Cl·(式(7)),Cl·也具有较强的氧化能力,可以降解ATZ,从而提高了ATZ的去除速率.而随着Cl-浓度的增加,Cl·会继续与Cl-反应生成Cl· -2(式(8)),而Cl· -2的氧化能力相对较弱,从而导致ATZ的去除速率降低(Buxton et al., 1999; Liang et al., 2006).
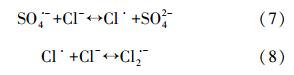
 |
| 图 6 Cl-(a)和CO2-3(b)对ATZ降解的影响(60 ℃,pH=7.0,[ATZ]0=50 μmol · L-1,[S2O2-8]0=1.0 mmol · L-1) Fig. 6 Effect of Cl-(a) and CO2-3(b)on the degradation of ATZ by persulfate(60 ℃,pH 7.0,[ATZ]0=50 μmol · L-1,[S2O2-8]0=1.0 mmol · L-1) |
CO2-3的存在也显著抑制了ATZ的降解,且抑制程度随着CO2-3的浓度的增加而增强(图 6b).当含有100 mmol · L-1 CO2-3 时,2 h后ATZ去除率只有40%,而没有CO2-3但其它条件相同时,ATZ已经完全去除.这可能是因为CO2-3会与SO· -4反应生成CO2-3(式(7)),而CO2-3的氧化能力很弱,所以会使ATZ去除率显著降低(Liang et al., 2006).
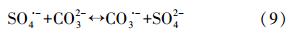
本文系统研究了利用热活化过硫酸盐降解水溶液中的ATZ,结果发现,升高温度或增加过硫酸盐浓度,ATZ的降解显著加快,热活化过硫酸盐降解ATZ遵循二级反应动力学规律;酸性和中性条件下ATZ的降解效率要高于碱性条件.通过自由基灭活实验可知,酸性和中性溶液中,起主导作用的自由基是SO· -4,而碱性条件下OH ·是主要的活性成分;HA和CO2-3的存在对ATZ的降解有抑制作用;低浓度的Cl-对ATZ的降解有促进作用,高浓度的Cl-则有抑制作用.这些因素,尤其是其它无机、有机物质对ATZ降解的影响在其它活化方式中同样存在,因此,在评价利用过硫酸盐降解ATZ的可行性,以及设计相关工艺时必须充分考虑.
| [1] | Anipsitakis G P,Dionysiou D D.2004.Radical generation by the interaction of transition metals with common oxidants[J]. Environmental Science and Technology, 38(13): 3705-3712 |
| [2] | Anipsitakis G P, Dionysiou D D, Gonzalez M A. 2006. Cobalt-mediated activation of peroxymonosulfate and sulfate radical attack on phenolic compounds. Implications of chloride ions[J]. Environmental Science and Technology, 40(3): 1000-1007 |
| [3] | Barreiro J C, Capelato M D, Martin-Neto L, et al. 2007. Oxidative decomposition of atrazine by a Fenton-like reaction in a H2O2/ferrihydrite system[J]. Water Research, 41(1): 55-62 |
| [4] | Bennedsen L R, Muff J, Sogaard E G. 2012. Influence of chloride and carbonates on the reactivity of activated persulfate[J]. Chemosphere, 86(11): 1092-1097 |
| [5] | Buster H R. 1990. Atrazine and other s-trazine herbicides in lakes and rains in Swizerland[J]. Environmental Science and Technology, 24: 1040-1058 |
| [6] | Buxton G V, Bydder M, Salmon G A. 1999. The reactivity of chlorine atoms in aqueous solution-Part II. The equilibrium SO4·-+Cl-ClNsbd +SO42[J]. Physical Chemistry Chemical Physics, 1(2): 269-273 |
| [7] | Chan K H, Chu W. 2006. Model applications and intermediates quantification of atrazine degradation by UV-enhanced Fenton process[J]. Journal of Agricultural and Food Chemistry, 54(5): 1804-1813 |
| [8] | Fang J Y, Shang C. 2012. Bromate formation from bromide oxidation by the UV/Persulfate process[J]. Environmental Science and Technology, 46(16): 8976-8983 |
| [9] | Freitas L G, Singer H, Müller S R, et al. 2008. Source area effects on herbicide losses to surface waters-A case study in the Swiss Plateau[J]. Agriculture, Ecosystems and Environment, 128(3): 177-184 |
| [10] | Gayathri P, Dorathi R P J, Palanivelu K. 2010. Sonochemical degradation of textile dyes in aqueous solution using sulphate radicals activated by immobilized cobalt ions[J]. Ultrasonics Sonochemistry, 17(3): 566-571 |
| [11] | Ghauch A, Tuqan A M. 2012a. Oxidation of bisoprolol in heated persulfate/H2O systems: Kinetics and products[J]. Chemical Engineering Journal, 183: 162-171 |
| [12] | Ghauch A, Tuqan A M, Kibbi N. 2012b. Ibuprofen removal by heated persulfate in aqueous solution: A kinetics study[J]. Chemical Engineering Journal, 197: 483-492 |
| [13] | Huang K C, Couttenye R A, Hoag G E. 2002. Kinetics of heat-assisted persulfate oxidation of methyl tert-butyl ether (MTBE)[J]. Chemosphere, 49(4): 413-420 |
| [14] | Kambhu A, Comfort S, Chokejaroenrat C, et al. 2012. Developing slow-release persulfate candles to treat BTEX contaminated groundwater[J]. Chemosphere, 89(6): 656-664 |
| [15] | Liang C J, Wang Z S, Mohanty N. 2006. Influences of carbonate and chloride ions on persulfate oxidation of trichloroethylene at 20 ℃[J]. Science of the Total Environment, 370(2/3): 271-177 |
| [16] | Liang C J, Su H W. 2009. Identification of Sulfate and Hydroxyl Radicals in Thermally Activated Persulfate[J]. Industrial and Engineering Chemistry Research, 48(11): 5558-5562 |
| [17] | Sene L, Converti A, Secchi G, et al. 2010. New aspects on atrazine biodegradation[J]. Brazilian Archives of Biology and Technology, 53(2): 487-496 |
| [18] | Mackul'ak T, Prousek J, vorc L. 2011. Degradation of atrazine by Fenton and modified Fenton reactions[J]. Monatshefte Fur Chemie, 142(6): 561-567 |
| [19] | Ma J, Graham N J D. 1999. Degradation of atrazine by manganese-catalysed ozonation: Influence of humic substances[J]. Water Research, 33(3): 785-793 |
| [20] | Niu C G, Wang Y, Zhang X G, et al. 2012. Decolorization of an azo dye Orange G in microbial fuel cells using Fe(II)-EDTA catalyzed persulfate[J]. Bioresource Technology, 126: 101-106 |
| [21] | Pennington D E, Haim A. 1968. Stoichiometry and mechamism of the chromium(Ⅱ) peroxydisulfate reaction[J]. Journal of the American Chemical Society, 90(14): 3700-3704 |
| [22] | Shih Y J, Putra W N, Huang Y H, et al. 2012. Mineralization and deflourization of 2, 2, 3, 3-tetrafluoro-1-propanol (TFP) by UV/persulfate oxidation and sequential adsorption[J]. Chemosphere, 89(10): 1262-1266 |
| [23] | Tauber A, von Sonntag C. 2000. Products and kinetics of the OH-radical-induced dealkylation of atrazine[J]. Acta Hydrochimica Et Hydrobiologica, 28(1): 15-23 |
| [24] | 王子键,吕怡冰,王毅,等. 2002.淮河水体取代苯类污染及其生态风险[J].环境科学学报, 22(3): 300-304 |
| [25] | Westerhoff P,Mezyk S P,Cooper W J,et al.2007.Electron pulse radiolysis determination of hydroxyl radical rate constants with Suwannee river fulvic acid and other dissolved organic matter isolates[J]. Environmental Science and Technology,41(13): 4640-4646 |
| [26] | Wu J,Zhang H,Qiu J.2012.Degradation of Acid Orange 7 in aqueous solution by a novel electro/Fe2+/peroxydisulfate process[J]. Journal of Hazardous Materials, 215-216: 138-145 |
| [27] | 杨照荣,崔长征,李炳智,等.2013.热激活过硫酸盐降解卡马西平和奥卡西平复合污染的研究[J].环境科学学报,33(1): 98-104 |
| [28] | 叶常明,雷志芳,王杏君,等. 2001.除草剂阿特拉津的多介质环境行为[J].环境科学,22(2): 69-73 |
| [29] | Zhao L, Deng Y R, Du Y X, et al. 2012. Study on the degradation of atrazine in photo-Fenton-like system under visible light irradiation promoted by N-doped Ta2O5[J]. Environmental Science, 33(4): 1252-1259 |