2.1 Quasi-Harmonic ApproximationThe free energy of Al was first calculated by the quasi-harmonic approximation and then corrected by taking into account the phonon-phonon interactions. The quasi-harmonic approximation (QHA) describes volume-dependent thermal effects based on lattice vibration models known as phonon, neglecting the phonon-phonon interactions. In the framework of the QHA, the Helmholtz free energy of a crystal system is,
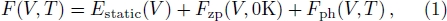
where
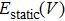
is the zero-temperature energy of a static lattice with volume
V. The term
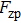
is the zero-point motion energy written as,
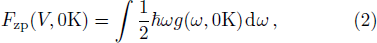
where
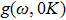
is the 0 K phonon density of states (PDOS) of the frequency
ω.The term
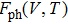
in Eq. (
1) is the lattice vibrational free energy, and is calculated from,
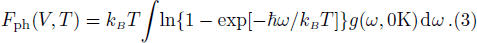
2.2 Anharmonicity CorrectionsIn the harmonic approximation, the PDOS 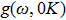 is calculated from the force constants using either the direct method[13–14] or the density functional perturbation theory.[5,15] In the harmonic approximation, the phonon-phonon interaction contribution to free energy are neglected, resulting in large errors of thermodynamic properties at high temperature. Therefore, the phonons interactions must be fully taken into account to include the rest of the anharmonic effects. However, the rest anharmonic free energy calculation is very challenging. As we know that the explicit anharmonicity are naturally implied in molecular dynamics (MD) simulations. We can take into account the rest anharmonic effects from MD simulations. The PDOS at T,
is calculated from the force constants using either the direct method[13–14] or the density functional perturbation theory.[5,15] In the harmonic approximation, the phonon-phonon interaction contribution to free energy are neglected, resulting in large errors of thermodynamic properties at high temperature. Therefore, the phonons interactions must be fully taken into account to include the rest of the anharmonic effects. However, the rest anharmonic free energy calculation is very challenging. As we know that the explicit anharmonicity are naturally implied in molecular dynamics (MD) simulations. We can take into account the rest anharmonic effects from MD simulations. The PDOS at T, 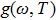 , can be extracted from the Fourier transform of autocorrelation of atomic position function
, can be extracted from the Fourier transform of autocorrelation of atomic position function 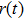 in a constant temperature MD simulation.[16]
in a constant temperature MD simulation.[16]
According to the Wiener–Khintchine theorem,[17–18] the autocorrelation of position function 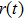 is given by
is given by
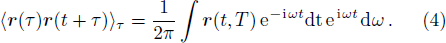
The PDOS at T is,
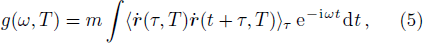
where
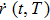
is the time derivative of the position
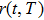
and
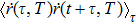
is the velocity autocorrelation in the MD simulation.
The Helmholtz free energy of a crystal system is then rewritten as,
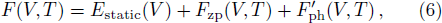
where
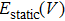
is the total energy of a static lattice withvolume
V.
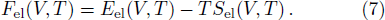
In Eq. (6), the term 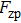 is the zero-point motion energy of the lattice written as,
is the zero-point motion energy of the lattice written as,
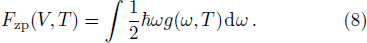
The term 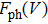 in Eq. (6) is calculated from,
in Eq. (6) is calculated from,
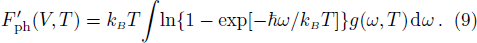
After obtaining the finite number of temperature dependent PDOS (TD-PDOS) data, we use a much strait-forward and simple method named the direct free energy interpolation (DFEI) method, to construct full anharmonic effects at any temperature.
The DFEI method extracts anharmonic free energy at any temperature based on the limited several TD-PDOS data from MD simulations. The free energies 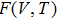 can be directly deduced from the TD-PDOS data at the specific temperatures Ts with the temperature interval 50 K, according to Eq. (6). The free energies at any temperatures were obtained by interpolating these resulting free energy data using the numerical analysis technique of the basis spline (B-spline). Finally, the anharmonic effects of other thermodynamic properties were corrected.
can be directly deduced from the TD-PDOS data at the specific temperatures Ts with the temperature interval 50 K, according to Eq. (6). The free energies at any temperatures were obtained by interpolating these resulting free energy data using the numerical analysis technique of the basis spline (B-spline). Finally, the anharmonic effects of other thermodynamic properties were corrected.
2.3 The Interatomic Potential of AlWe perform classical MD simulations to simulate Al at different pressure and temperature. Then, we gather the trajectories of all the atoms in crystal to calculate phonon density of states. The embedded-atom-method (EAM)[19–20] potential developed by Mishin et al.[21–22] was used to describe the interatomic interactions of Al atoms. For a metal system containing N atoms, the total potential energy 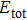 is the sum of the embedding energy F and a pair potential ϕ,
is the sum of the embedding energy F and a pair potential ϕ,
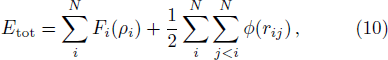
where
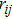
is the distance between the atoms
i and
j. The function
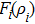
is the energy to embed the atom
i into the background electron density
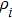
, which is the superposition of the atomic densities,
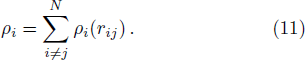
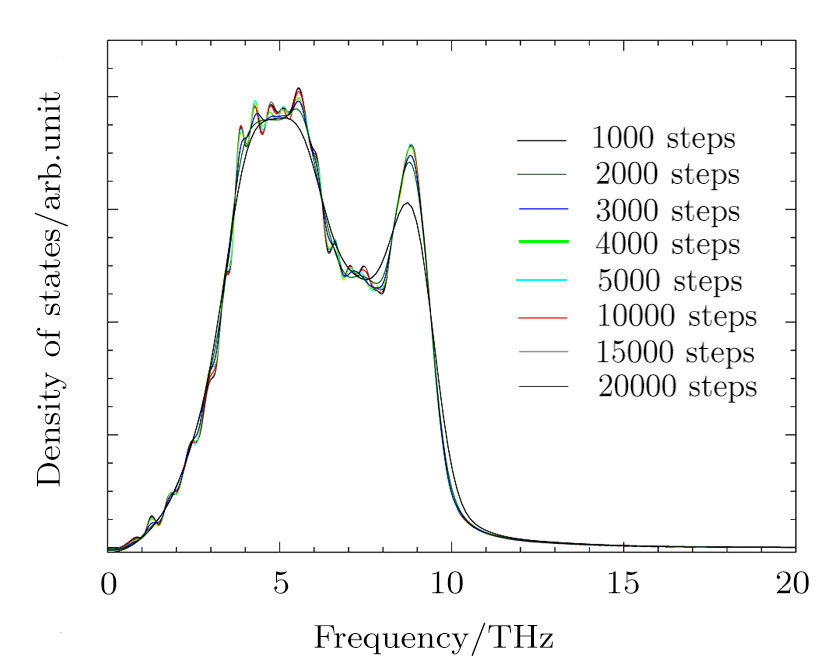
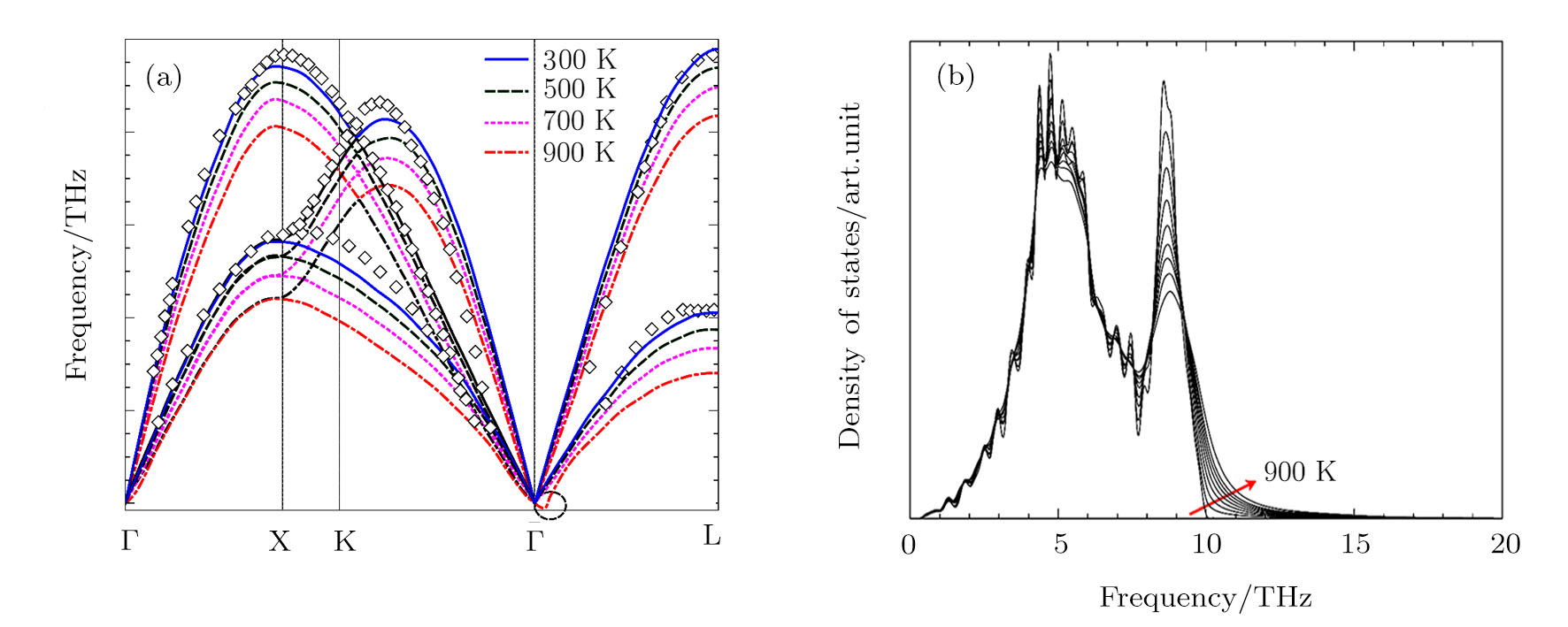
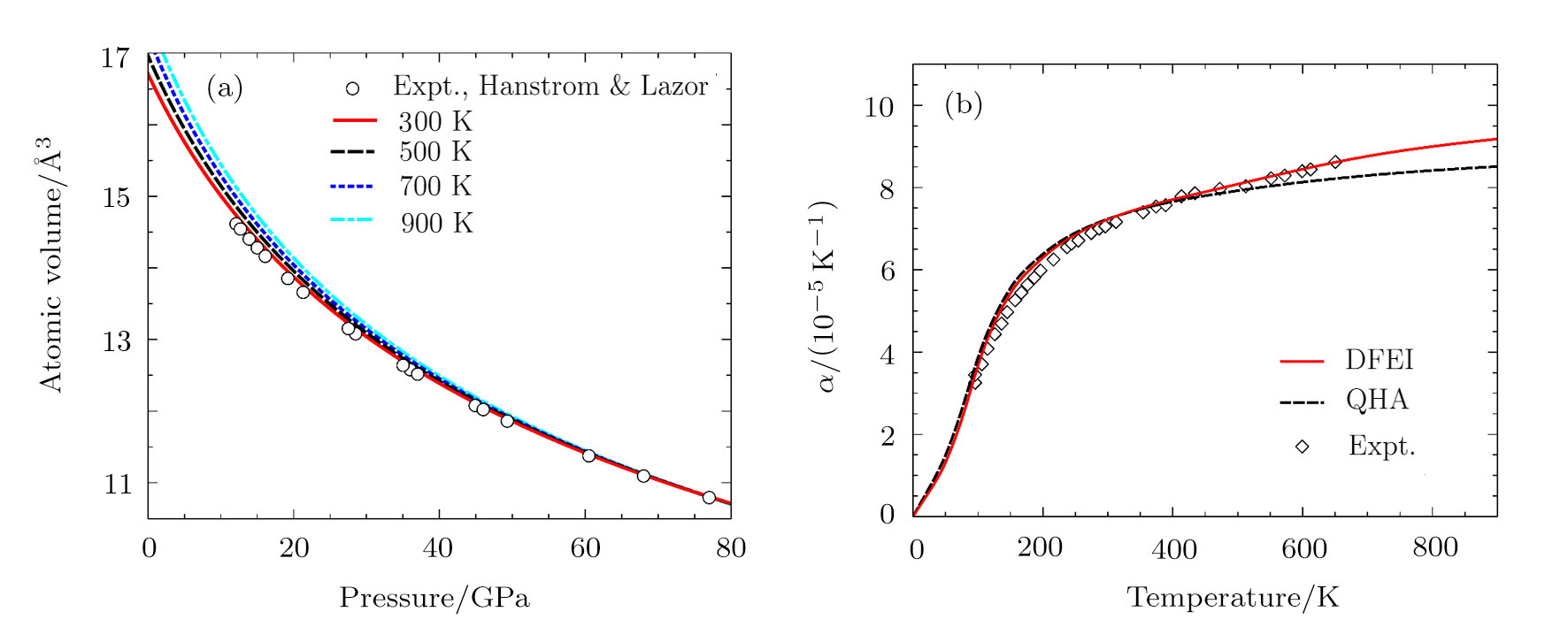
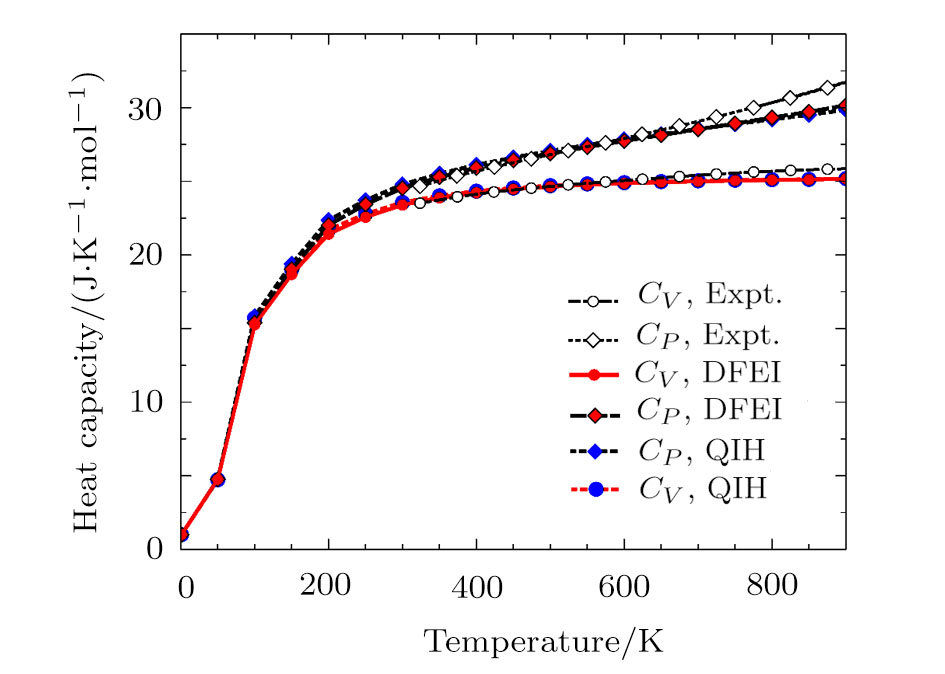
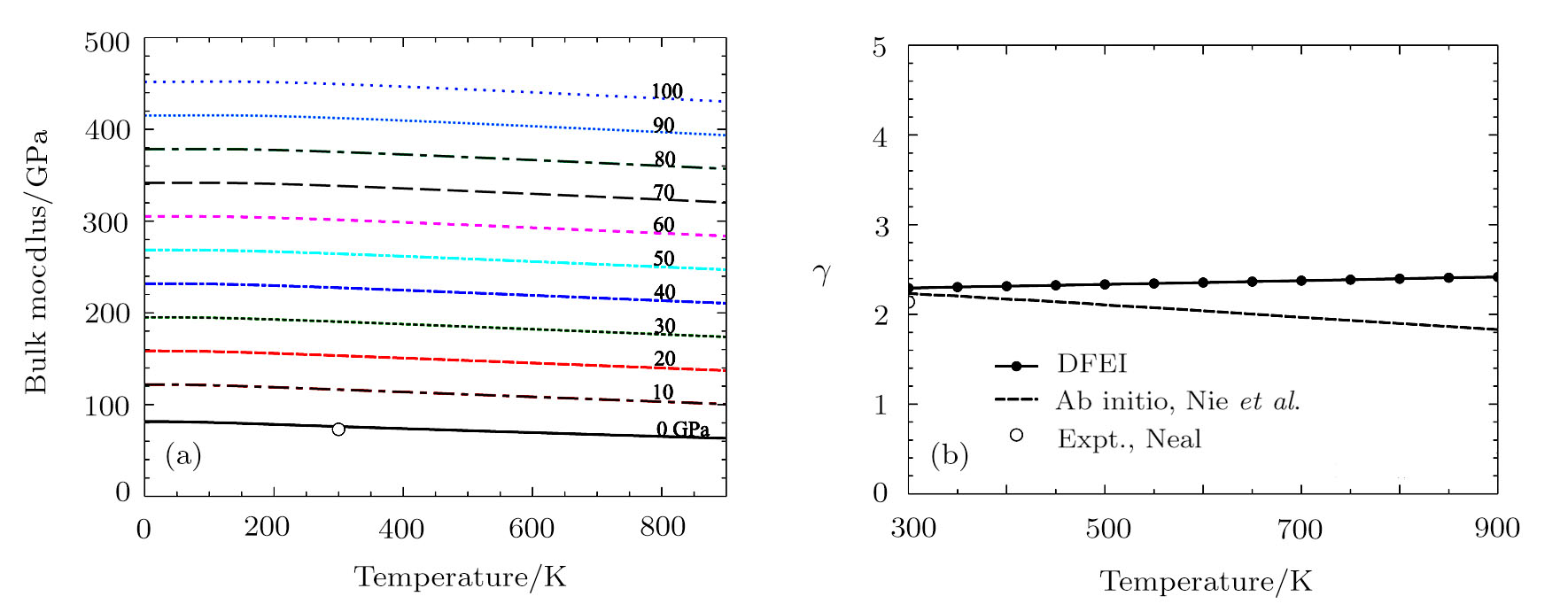
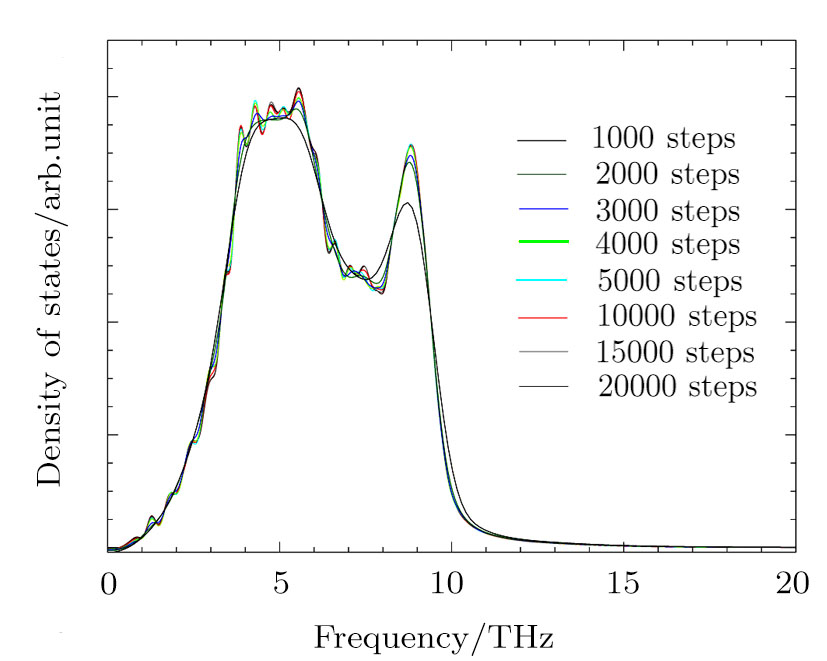
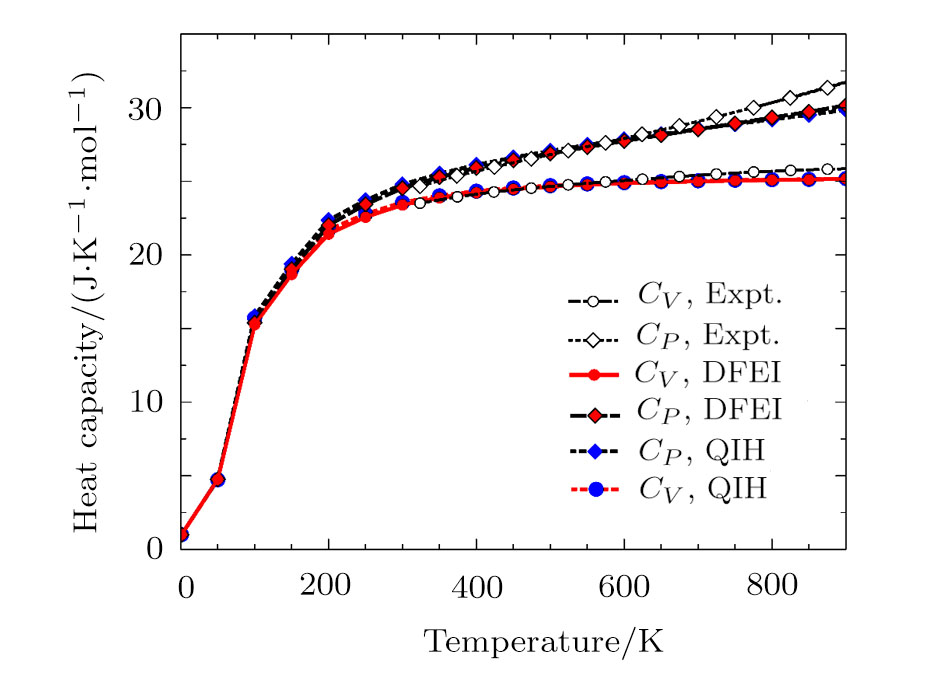
The MD simulations were conducted with the large-scale atomic/molecular massively parallel simulator (LAMMPS)[23–24] package. The simulation box constructed from the multiplication 14×14×14 of the face-centered-cubic (fcc) conventional unit cell including 10976 atoms. The simulations were conducted for all the supercells with different volumes in the canonical ensemble (NVT). The periodic boundary condition was used for all the atoms in the simulation box. The time step was 1 fs and the total number of time steps were 10000. From Fig. 1, we see that 10000 MD steps are able to converge the PDOS well.


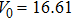
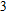
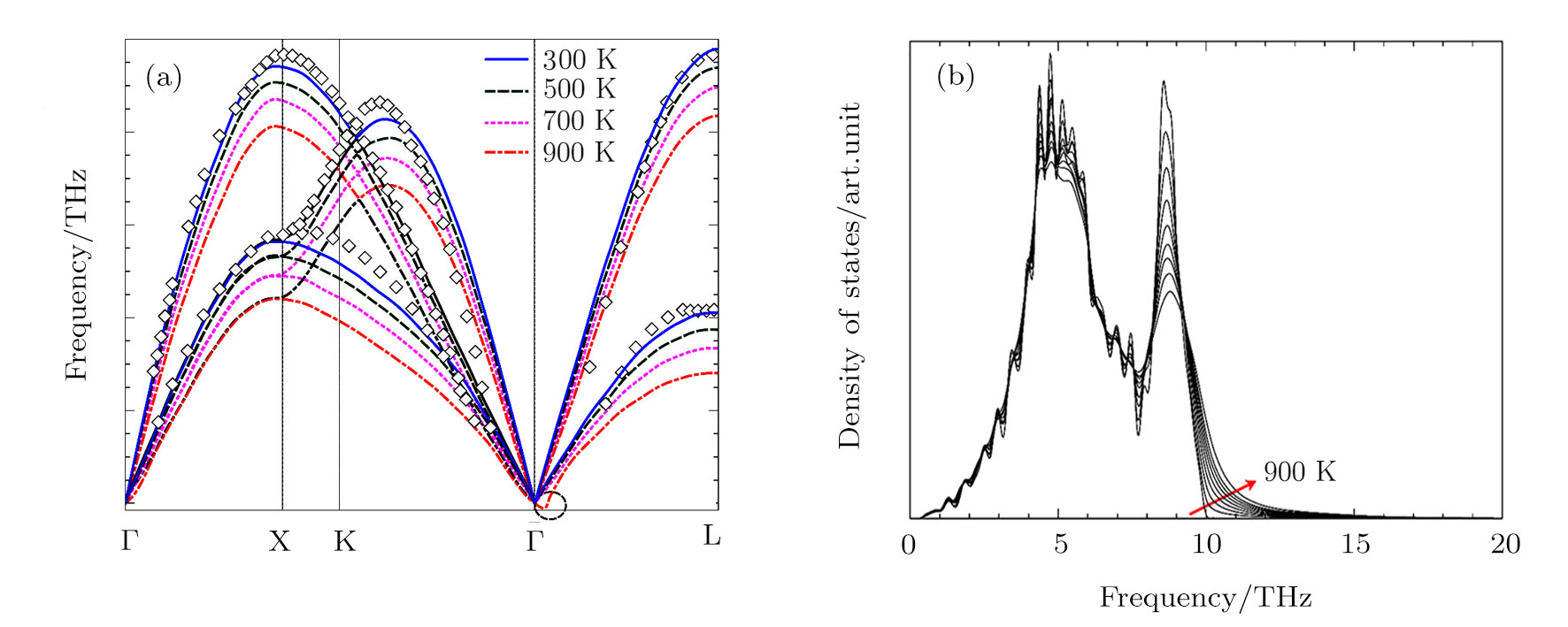
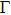
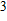
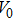
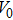
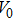
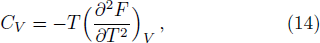
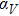
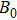
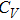
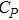
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}