 2015, Vol. 33
2015, Vol. 33扩展功能
文章信息
- 李飞, 武思琴, 刘柯
- LI Fei, WU SiQin, LIU Ke
- 鲕粒原生矿物识别及对海水化学成分变化的指示意义
- Identification of Ooid Primary Mineralogy:A clue for understanding the variation in paleo-oceanic chemistry
- 沉积学报, 2015, 33(3): 500-511
- ACTA SEDIMENTOLOGICA SINCA, 2015, 33(3): 500-511
- 10.14027/j.cnki.cjxb.2015.03.008
-
文章历史
- 收稿日期:2014-04-28
- 收修改稿日期:2014-07-19



2. 中国地质大学(武汉)生物地质与环境地质国家重点实验室 武汉 430074
2. State Key Laboratory of Biogeology and Environmental Geology, China University of Geosciences, Wuhan 430074
鲕粒是由钙质纹层围绕单个核心而形成的圆形或椭圆形包覆颗粒(coated grain),粒径一般小于2 mm,以光滑、均匀的纹层结构而与核形石、渗流豆粒等相区分。从Sorby于1879年对鲕粒展开详细调查至今[1],研究者们对鲕粒的形貌结构[2, 3]、沉积背景[3, 4]、古气候意义[5, 6]、烃类储层[7]等方面开展了大量卓有成效的工作,极大地推动了人们对于这种“迷人的”碳酸盐颗粒的认识。但在鲕粒成因这个最基本的问题上争议不断,到目前为止较为大多数人所接受的“鲕粒化学成因”观点[8],正受到越来越多微生物化学证据的挑战[9, 10, 11, 12, 13, 14, 15]。隐藏在这个基本问题之下的鲕粒矿物组成问题则很少见于国内文献,关于鲕粒成岩特征的研究也对该问题进行了回避。虽然鲕粒原生矿物的组成及控制因素比较复杂,但却影响了鲕粒的纹层结构、保存程度、成岩特征,并且能够提供指示海水化学成分变化的线索,意义重大。基于此,本文从鲕粒原生矿物的组成入手,回顾了前人对该问题的认识过程,综述鲕粒原生矿物的识别特征,对原生矿物识别在成岩特征、古海洋化学上的意义进行阐述并探讨。
1 鲕粒原生矿物与结构研究进展虽然Sorby[1]在研究鲕粒之初已经认识到存在一部分方解石质的鲕粒能够保存下来原生的结构和矿物成分,但直到上个世纪70年代初,人们一直认为古代鲕粒和现代海洋环境下“巴哈马”型鲕粒一样,均由文石矿物组成。对于当时发现的一些具有放射状纹层结构的鲕粒则认为是文石质鲕粒经过成岩作用改造的结果[16, 17] 。Shearman等人[18]在研究波斯湾特鲁西尔海岸(Trucial Coast)鲕粒时,更是提出切线状结构的保存有赖于纹层间的有机质夹层(organic template)保护,放射状结构的出现与有机质夹层缺失有关。这种观点遭到Kahle[19]、Sandberg[20]和Simone[21]等人的强烈质疑。Sandberg[20]在对现代大盐湖(Great Salt Lake)具放射状纹层结构的文石鲕以及石炭系密西西比亚系保存有放射状精细结构的鲕粒进行重新研究后认为,放射状纹层结构是沉积作用形成的原生结构,并非是成岩阶段文石发生稳定转换之后的结果(图 1)。之后,Marshall和Davies[22]、Milliman和Barretto[23]、Davies和Martin[24]、Halley[25]以及Medwedeff和Wilkinson[26]等也发现多个现代高镁方解石质鲕粒具有原生放射状结构的实例。在晚寒武世[27],中、晚侏罗世[28] ,早白垩世[21]等均发现有大量结构保存完好的放射状、同心—放射状低镁方解石质鲕粒。在不同地质历史时期,鲕粒纹层结构和保存程度的差异证实鲕粒原生矿物类型并非一成不变,存在包括文石、低镁方解石和高镁方解石在内的三种主要形式。

之后,研究者们对这些原生放射状结构的成因再次陷入争论。其中一类观点认为鲕粒放射状与切线状结构的差异与形成环境水动力条件有关[27, 29, 30]。这种观点认为鲕粒切线状纹层结构形成于强水动力条件之下,而放射状纹层结构形成于水体能量相对较弱的环境中。因此认为具有切线状和放射状纹层的鲕粒形成于两种不同的水动力条件之下。这类观点得到较多研究实例的支持,至今仍有相当多的研究者坚持该观点。然而,原生单矿物方解石质鲕粒绝大多数都具有放射状纹层结构(陆相渗流成因鲕粒除外),把众多方解石质鲕粒全部归于水体能量较低的环境显然不太合适。目前较为确定的是文石质的放射状纹层要比文石质的切线状纹层形成水体能量低;另一类观点认为盐度与纹层结构关系更为密切[23, 25, 31]。这类观点认为原生放射状结构鲕粒形成于盐度异常(咸水或淡水)环境下,而切线状纹层更容易在正常盐度海水环境下形成。盐度的异常促使鲕粒原生结构发生改变。但也存在相当多的实例表明正常盐度海水环境下同样可以发育大量的放射状结构鲕粒[22, 24, 28]。
Strasser[32]甚至综合水动力条件与盐度这两类观点后提出了一种双因素控制模式,即在广海强水动力条件下更多地形成切线状纹层鲕粒,在间歇性动荡水体、中等盐度环境下以同心—放射状纹层结构为主,在静水、局限环境下(高盐度)以放射状纹层结构为主。虽然这种观点到目前为止仍存在较大影响,但无法忽视的是它在解释具体实例时显得力不从心。例如:①华北寒武系第三统至第四统广泛发育放射状鲕粒的同时,切线状结构鲕粒少见;②早三叠世华南广泛发育的鲕粒中几乎不含放射状结构;③同时发育切线状和放射状纹层结构的鲕粒集中在中、晚侏罗世,晚宾夕法尼亚世等少数时间段内,等等。由于当时普遍认为显生宙海水化学成分与现代海洋类似,对于鲕粒原生矿物成因的解释只是从局部小环境角度入手,未考虑能够控制纹层类型的总体变量。
Sandberg[20]和Wilkinson和Landing[28]在对不同时代鲕粒纹层结构和保存程度的研究中发现,在晚中生代至早新生代期间以结构保存较好的方解石质鲕粒为主,与现代海洋环境中的鲕粒存在明显不同,并推测当时的海水化学成分与现代存在差异。同时,Sandberg[20]提出鲕粒原生矿物类型与海水化学成分存在关联:即在富镁的海水中(Mg/Ca> 2),鲕粒原生矿物主要以文石、高镁方解石为主(镁离子对方解石生长具有抑制作用,且镁离子易于进入方解石晶格形成高镁方解石)[33],纹层类型包括切线状(文石鲕)和放射状(主要为高镁方解石鲕)两种;在贫镁的海水中(Mg/Ca<2),有利于低镁方解石的形成(具体机理目前仍不完全清楚,可能与方解石生长不再受到镁离子抑制有关),在这种条件下形成的鲕粒具有放射状或同心—放射状纹层(图 1B)。Sandberg[20]对鲕粒原生矿物类型及识别标志上具“里程碑”意义的研究推翻了人们近一个世纪关于鲕粒矿物组成的认识,极大地促进了鲕粒原生矿物、成岩特征和古海洋意义方面的研究。之后,Sandberg[34]通过对显生宙海相碳酸盐鲕粒原生矿物的深入研究,识别出三个“抑制方解石生长”的时期(早寒武世、晚石炭世—晚三叠世/早侏罗世、早/中新生代—现代),以及两个“利于方解石生长”的时期(中寒武世—早石炭世,晚三叠世/早侏罗世—早/中新生代。这两类不同海水化学条件所处的时期也被分别称作“文石海”和“方解石海”(图 2)。

由于文石和高镁方解石属于亚稳定性矿物,会受成岩作用的影响发生稳定转换。地层中保存的古代文石鲕和高镁方解石鲕粒,已几乎完全被低镁方解石取代。因此,恢复古代鲕粒的原生矿物组成就显得非常重要。 2 鲕粒原生矿物识别特征
对原生矿物为文石的古代鲕粒,Sandberg[34]按识别标志的可靠性程度由高到低区分如下:①仍保存为文石质(最为可信的标志,但是古代实例非常稀少);②粗晶方解石镶嵌结构(已方解石化),通常原生定向的文石结构已被破坏,但仍具有部分残余结构;③不含文石结构残余的方解石镶嵌结构,但Sr浓度非常高(一般在2 500 ppm以上);④既不含文石结构残余也不具备高浓度Sr的方解石镶嵌结构;⑤被胶结物充填的鲕粒铸模孔(文石鲕更容易形成铸模孔,但无法排除低镁方解石或高镁方解石质鲕粒受成岩作用影响发生溶解的可能);⑥保留有精细的纹层结构(推测为方解石化的文石假晶)。其中①和②的可信度最高,③至④的可信度有所降低,如果保留文石成岩特征的证据则可信度仍非常高,⑤和⑥的可靠性较低,需要其他更可靠的证据来进一步地确定。
概括起来,判断鲕粒原生矿物类型主要依靠:①原生纹层结构;②保存程度;③微量元素浓度,尤其是Sr-Mg的浓度。
2.1 原生纹层结构现代开放海洋环境中形成的文石鲕与方解石鲕具有不同的纹层结构(图 3),这为区分古代鲕粒原生矿物提供了重要基础[21, 26]。

|
| 图 3 海相环境下单矿物碳酸盐鲕粒纹层结构与原生矿物对应关系 Fig. 3 Comparison between cortical fabrics and original mineralogy of monomineralic ooids in marine settings |
海相文石质鲕粒具有细的纹层、微米级的晶体沿鲕粒外表面的切线方向定向排列。现代海相环境中文石质鲕粒绝大多数为切线状结构[38],局限海洋环境以及非海相环境中经常可以看到具有放射状结构的文石鲕[20, 24, 31, 39, 40]。在这些例子中,大多数是受异常水体环境(如超盐度或是微咸水)干扰造成的,但也的确存在正常海洋盐度水体中发育放射状文石鲕的报道(来自澳大利亚东北部海滩[24])。 2.1.2 方解石质鲕粒
方解石质鲕粒通常具有放射状或者同心—放射状结构,纹层内粗晶晶粒(50~500 μm)呈辐射状从鲕粒核心向外延伸[22, 23]。从对已有文献整理的情况来看,无论古代还是现代海相环境下形成的鲕粒,目前为止均未见切线状低镁方解石鲕粒的报道[3, 21, 26, 41],仅有的实例出现在陆相钙结岩中[3]。
2.1.3 双矿物鲕粒(bimineralic ooids)双矿物鲕粒是指纹层由两种不同矿物构成(主要为文石+高镁方解石,少量为文石+低镁方解石)的鲕粒,包括方解石质内层与文石质外层组合,或者文石和方解石质纹层相间排列结构(图 1C,D)[38, 42, 43, 44, 45, 46]。现代双矿物鲕粒在海相[44, 47]或者湖相环境[48]中均有报道。其中,文石质纹层晶体排列方式常常表现为切线状,而低镁方解石[48]或高镁方解石质纹层[47]表现为放射状。对于相间排列结构的双矿物鲕粒,其纹层晶体排列方式目前还不是非常清楚。而对于双矿物鲕粒的形成原因,目前尚无统一认识,可能与水动力条件[27, 30, 47],区域环境内Mg/Ca的变化[48],或者海水饱和度的变化存在关联[49]。可以确定的是水动力条件不会是双矿物鲕粒形成的主要控制因素。如果水体能量是双矿物鲕形成控制因素的话,在地质历史时期应该存在众多的双矿物鲕粒,而不是像现在地层中发现的一样,集中在特定地质时期内。湖泊、澙湖等易受低Mg/Ca的淡水影响,小环境内Mg/Ca的波动可能会影响鲕粒的原生矿物成分;但在开放海洋环境下,Mg/Ca主要受洋中脊热液活动缓慢控制且海水混合速率非常高,不太可能出现迅速波动的情况。
2.2 保存程度文石和高镁方解石在埋藏环境下由于热力学不稳定性而发生溶解,或者重结晶转化为稳定的低镁方解石[38]。重结晶后通常转化为由细粒至粗粒亮晶方解石组成的镶嵌结构,这种转化将会破坏原生纹层结构,造成指示晶体定向性特征难以保存下来。同时,溶解导致鲕粒形成溶孔,也有可能重结晶后形成铸模孔。作为对比,原生矿物为低镁方解石的鲕粒通常能够抵抗成岩作用的影响,纹层结构和晶体定向性特征能够得以保存。现将文石鲕、方解石鲕和双矿物鲕的保存特征介绍如下:
2.2.1 文石质鲕粒鲕粒原生矿物从文石转变为低镁方解石后,原生结构会遭到不同程度破坏,从结构保存相对完好的新生变形鲕粒到鲕粒铸模孔,甚至仍保留为溶孔的情况均可能发生[42, 43],这与成岩系统的封闭程度、有机质成分的阻隔等存在关联。文石质鲕粒虽然易受到成岩作用的改造,但保留下来的特殊溶解、破碎结构能够帮助我们进行更好地识别,其中包括砖砌结构(brickwork structure),溶解变形结构,偏心结构(displaced structure)等(图 4A,C、图 5)。

|
|
图 4 判断鲕粒原生矿物类型的典型结构 A.砖砌结构,指示文石质原生矿物成分;B.剥落纹层,由于内部纹层发生溶解,内外压力不平衡而发生破碎剥落,推测鲕粒原生结构可能为双矿物成分;C.偏心结构鲕粒,指示文石质纹层溶解后,鲕核受重力影响发生偏移;D.偏心鲕粒,与C在成因、形成环境等方面存在显著差别,对鲕粒原生矿物的指示意义不大;E.一类特殊的双矿物鲕粒,内皮层为小的放射状鲕粒(或不发育内皮层,文石质外皮层直接与鲕核接触),外皮层溶解后被放射状方解石质内皮层(左侧)或鲕核(右侧)的共轴柱状胶结物取代(也可能与放射状文石质纹层溶解后的残余结构有关);F.被基质填充的放射状结构,基质为黑色,沿鲕核呈辐射状排列。推测其原生矿物为(高镁)方解石。 Fig. 4 Diagnostic cortical fabrics for identifying ooid primary mineralogy |

|
|
图 5 原生文石质鲕粒镜下特征 A.切线状纹层结构。寒武系第三统,河北邯郸;B.白云岩化鲕粒(箭头所指),白云石颗粒显示出同心状排列方式(箭头方向)。寒武系第三统,北京西山;C.鲕铸模孔(A)与“鸭形”鲕(B)。下三叠统,广西平果;D.鞍状白云石鲕(A)与柱状结构鲕粒(B)。柱状结构鲕粒中方解石呈锥形,尖端指向核心,钝端平直,呈辐射状排列。晶体表面具双晶纹或菱形解理,显示重结晶特征。寒武系第三统,河北邯郸。 Fig. 5 Photomicrographs of formerly aragonitic ooids,plane-polarized light |
砖砌结构是指鲕粒纹层发生新生变形作用后形成的次生方解石以砖块叠加的方式,围绕核心构成的同心状结构。文石的溶解被严格限制在单个纹层内,纹层之间的界限与原结构基本保持一致。砖砌结构目前已经被广泛应用于对文石鲕的判别[43, 46, 50, 51, 52],但对于其成因目前还不太清楚,推测可能与层间微生物结构的阻隔有关[50] 。
溶解变形结构是指在成岩作用影响下形成的一系列选择性溶解结构(图 5C),包括具有鸭形(duck-like structure)[53]、象鼻—象尾相连形(elephant trunk-to-tail texture)[54]和链形(zigzag chains)[55]等结构的扭曲鲕(distorted ooids),显示鲕粒在溶解和早期胶结过程中发生形变,指示其原生矿物很可能为文石。
偏心结构是指鲕粒文石质纹层发生溶解后,非文石质鲕核在重力作用下发生偏移所形成的结构(图 4C)。需要注意的是偏心结构与偏心鲕粒(asymmetrical ooids/eccentric ooids;指核心在生长过程中就已发生迁移;图 4D)概念和意义上的差别。
2.2.2 方解石质鲕粒方解石质鲕粒以镁含量的不同(以4 mol % MgCO3为界限)分为低镁方解石鲕和高镁方解石鲕两种。低镁方解石为稳定矿物,在大多数成岩环境下低镁方解石质鲕粒都可以保留原生结构、微量元素信息[56],而高镁方解石为亚稳定矿物,经成岩作用转换为更稳定的低镁方解石。但这个过程中高镁方解石常常会保留下来原生结构(保存原生晶格排列方向的假晶)。这样低镁方解石与高镁方解石鲕粒均保留有放射状或同心—放射状结构,这与发生溶解的文石鲕区分明显(图 6A,B)。

|
|
图 6 方解石质鲕粒和双矿物鲕粒镜下特征 A.同心—放射状结构方解石质鲕粒。寒武系第三统,河北邯郸;B.微白云石颗粒(箭头),推测其原生矿物为高镁方解石。寒武系第三统,河北邯郸;C.双矿物鲕粒,内皮层为保存良好的方解石质放射状结构,外皮层为残余文石质切线状结构。寒武系第三统,北京西山;D.双矿物鲕粒背散射图像(电子探针测试显示深色纹层具有较高的Mg含量)。下三叠统,广西平果。 Fig. 6 Photomicrographs of calcitic and bimineralic ooids,plane-polarized light |
两种方解石质鲕粒从原生结构上看,差别不明显,较难区分。但在封闭成岩系统下,高镁方解石鲕在发生稳定转换过程中,镁元素从方解石晶格中流失,常以微白云石包裹体(microdolomitic inclusions)的形式嵌入到低镁方解石纹层中[57],可以通过偏光显微镜或扫描电镜进行识别(图 6B)。同时,高镁方解石鲕粒在早期成岩过程中容易受到铁的污染(镁方解石溶解,混入指示成岩作用的铁离子后形成含铁方解石),与不易受成岩作用影响的低镁方解石鲕粒可以通过添加铁染色剂(加铁氰化钾与茜素红混合溶液显示蓝紫色)[55, 58]的方式进行区分。
2.2.3 双矿物鲕粒双矿物鲕粒可以通过对比纹层结构和保存程度的差异进行原生矿物识别。其中文石质纹层受成岩作用影响,原生结构发生溶解,一般保存为镶嵌结构或砖砌结构;方解石质纹层仍然保留较为完整的放射状原生结构(部分高镁方解石质纹层可能发生溶解)(图 6C)。另外,不同矿物组分的纹层受成岩作用影响的程度存在差异,可以利用两类纹层在阴极发光特征上的差异进行判别[44];或者通过对比扫描电镜下背散射图像进行矿物成分区分(图 6D)。
当双矿物鲕粒中文石质纹层发生溶解形成空腔时,部分双矿物鲕粒会因内外压力不平衡而发生破碎,形成剥落纹层(spalled cortices)。剥落的方解石质纹层由于文石质成分的缺失而难以将形貌完整拼合,而且核心常常偏移中心位置(图 4B)。这与鲕粒受埋藏、压实作用产生的机械破碎在成因和结构上完全不同,因而可以将剥落纹层作为双矿物鲕粒鉴定的依据之一。
2.3 特殊鲕粒结构 2.3.1 不常见的识别特征除上述较为常见的鲕粒纹层结构和保存特征外,还存在一些可以指示原生矿物成分的特殊结构:
(1) 其他矿物类型
包括石英、白云石等矿物充填鲕粒内部孔隙,指示鲕粒原生矿物很可能为文石,受成岩交代作用的影响,以微晶石英或鞍状白云石颗粒(图 5D)的形式充填鲕粒内部结构[51]。
(2) 柱状结构
鲕粒外皮层由辐射状排列的柱形方解石晶体构成。靠近柱形方解石外侧的位置常发生溶解,由微亮晶方解石充填。柱形方解石内侧(靠近鲕粒核心)与放射状纹层结构接壤(此种情况较多),或柱形方解石内侧直接与鲕核接触。柱状结构的成因可能与鲕粒外皮层溶解后被放射状方解石质内皮层的共轴柱状胶结物取代有关。需要注意的是,有一类方解石鲕粒具有亮晶—泥晶两种辐射状纹层(图 4F),与柱状结构的文石鲕相比粒径较小(平均粒径0.5 mm,最大0.7 mm),且黑色泥晶辐射纹层从核心延伸至鲕粒外表面,差别明显[51]。
2.3.2 具有争议性的保存结构(1) 镶嵌结构鲕粒和鲕铸模孔
虽然镶嵌结构鲕粒和鲕铸模孔(图 5C)经常与具典型文石特征的鲕粒共生[43, 54, 59],但由于其本身也可以在深埋藏成岩环境或成岩晚期大气渗流环境下形成[30],所以不能简单地将这两类归入文石鲕粒的鉴别特征。鲕粒原生矿物的识别还需要其他更为可信证据的支持。
(2) 微晶鲕粒(micritic ooids)
纹层由不超过10 μm的等粒状超微方解石晶粒构成。虽然不稳定矿物发生成岩转换过程中很容易形成微晶结构,且微晶鲕粒经常与新生变形作用形成的镶嵌结构鲕粒或鲕铸模孔共生,但不能忽视的是泥晶化的方解石鲕,或在微生物钻孔作用掩盖或者破坏下形成的微晶纹层结构,而且这些过程均是广泛存在的。微晶鲕粒作为鲕粒原生矿物识别标志的可靠性较镶嵌结构鲕粒或鲕铸模孔更低。
2.4 微量元素浓度文石质鲕粒与方解石质鲕粒在某些微量元素浓度上,也存在较大差别,其中最为重要的是Sr。Sr元素在文石和方解石质沉积物中的分配系数具有显著差异,分别为1.14和0.14[60](Kinsman,1969)。现代海洋环境下文石质沉积物中Sr浓度高达8 000~10 000 ppm[8, 38, 56, 60],而方解石质沉积物中Sr浓度平均只有1 500~2 000 ppm[60]。Sr元素浓度的明显差别为我们区分文石鲕和方解石鲕提供了重要线索(图 7)。这种差别也体现在古代鲕粒中,但要注意不同地质历史时期海水Sr浓度背景值并非完全一致,分析时需要考虑实际情况。

对古代鲕粒Sr浓度的判断必须考虑成岩作用的影响。在碳酸盐成岩过程中,微量元素浓度也会发生相应变化,通常Sr和Na会逐渐降低,而指示成岩过程的Fe和Mn浓度会逐渐增高[56]。一般通过1 000Sr/Ca与Mn、Fe对应关系,或Sr/Mn比值判断成岩系统的封闭程度。在封闭—半封闭成岩环境下,保存的文石质鲕粒中Sr浓度仍保持在1 000~5 000 ppm,而方解石质鲕粒中Sr浓度一般在1 000 ppm以下[43, 52, 59, 61, 62, 63];如果鲕粒保留有高的Sr(大于2 000 ppm)和低的Fe和Mn含量(一般在数百ppm),可以推测其原生矿物为文石,且受成岩作用影响较小(封闭成岩环境);如果Sr浓度不超过1 000 ppm,而Mn含量仍保持低值,需要考虑鲕粒原生矿物是否为方解石;如果鲕粒具有不一致的Sr浓度(从小于1 000 ppm至数千ppm)和与之成反比的Mn浓度,可以推测鲕粒受到成岩作用的改造,其原生矿物可能为文石(图 8)。需要注意的是:①高的Fe和Mn含量并非完全是成岩作用造成的结果(与水体中氧含量及沉积速率也存在关联);②在开放成岩环境下,鲕粒中Sr浓度不论高低,均不宜作为判断鲕粒原生矿物类型的地球化学标志。对于这些特殊情况,上述元素浓度判断的方法可能并不适合,需要结合其他标志才能进行原生矿物判断。

Mg在方解石中的浓度以MgCO3计,一般在0~18 mol %之间,超过20 mol% 浓度的镁方解石非常少见。受温度、饱和度等因素的影响,镁在方解石中的分配系数范围从0.01至0.08之间变动[4]。文石中Mg的分配系数只有0.000 16[64],而且文石几乎不含有超过1.5 mol %的MgCO[38, 43, 44]3。根据Mg含量的不同(以4 mol % MgCO3为界限)可以轻易区分出现代海洋环境中的低镁方解石与高镁方解石鲕粒。然而,在判断古代方解石质鲕粒原生成分时存在较大难度,这是由于:①受成岩系统封闭程度的影响,高镁方解石中Mg离子容易流失;②由于Mg在成岩流体中来源的多样性,次生沉淀下高浓度的Mg与原生高镁方解石中的Mg难以区分。我们需要在大量测试分析的基础上,根据鲕粒保存下来的MgCO3浓度,结合Sr浓度(一般在1 000 ppm以下),推测方解石质鲕粒属于低镁还是高镁方解石矿物。
受鲕粒保存程度及测试条件的限制,从单个因素出发往往只能推测鲕粒原生矿物类型,只有综合考虑原生纹层结构、保存程度以及微量元素浓度这三条原则,才能对鲕粒原生成分做出更为准确的判断。
3 鲕粒原生矿物类型控制因素地质历史时期鲕粒原生矿物成分的变化主要受海水化学条件的控制,尤其是二氧化碳分压[66],Mg/Ca比值[34]以及碳酸盐饱和度[67, 68]。
3.1 Mg/Ca与大气二氧化碳分压Sandberg[20]基于“水体中Mg/Ca的变化影响碳酸钙矿物结构的同质异相性”以及“中生代以来钙质超微生物及浮游有孔虫的繁盛造成海水中钙离子移除”,提出鲕粒原生矿物类型与海水Mg/Ca存在关联,即在海水高Mg/Ca时期主要形成文石质鲕粒,低Mg/Ca时期主要形成(低镁)方解石鲕粒。由于这种观点无法解释古生代海水化学特征,Sandberg[34]随后放弃了这种观点,转而认为鲕粒原生矿物类型与区域性构造运动造成的二氧化碳分压变化有关[66]。但Wilkinson等[42]认为海水Mg/Ca与大气二氧化碳分压这两种因素可能共同影响着鲕粒原生矿物类型。通过对比显生宙不同时期鲕粒原生矿物,Wilkinson等人[42]认为一级海平面变化处于高水位期时,洋中脊正处于扩张期,玄武岩热液蚀变吸收海水中的镁离子并释放钙离子,同时将产生的大量二氧化碳排入大气增加大气二氧化碳分压,对应低镁方解石质鲕粒形成时期;当一级海平面变化处于低水位期时,洋中脊扩张速率减弱,造成镁离子吸收作用也相应减弱。陆源物质暴露、风化剥蚀作用及光合作用增强消耗大量二氧化碳,大气二氧化碳分压降低,对应文石鲕和高镁方解石质鲕粒形成时期。
尽管大气二氧化碳分压在显生宙的波动趋势与海水Mg/Ca有着较好的负相关性(图 2),但其无法解释1)蒸发岩类在MgSO4型(对应“文石海”时期)和KCl型(对应“方解石海”时期)之间的转换;以及2)如何持续维持只形成方解石质鲕粒的“方解石超饱和而文石不饱和的海水化学条件”(详见3.2节)而受到较多的质疑[69, 70]。基于此,Hardie[35]以及Stanley和Hardie[71]对20世纪90年代以前的观点进一步修正,提出洋中脊产生速率的周期性变化控制着洋中脊热液活动的周期性变化,进而导致海水化学成分的波动。洋中脊玄武岩与海水反应形成绿岩的过程中释放钙离子和钾离子的同时吸收镁离子和硫酸根离子。高的洋中脊扩张速率(洋壳形成速率高的时期)导致钙离子浓度相对升高而镁离子浓度相对降低(低Mg/Ca时期),这与Sandberg[20, 34]通过非生物碳酸盐沉积物(鲕粒和胶结物)划分的“方解石海”时期相吻合。同时,这种观点得到蒸发岩类型[35]、流体包裹体[72]、生物礁及生物矿化[69, 71, 73]等一系列证据的支持,因而洋中脊产生速率被认为是长时间尺度下(100~200 Ma)海水化学成分波动的主要控制因素[71]。
3.2 海水碳酸盐饱和度海水中碳酸盐饱和度受到温度、盐度,以及二氧化碳分压的影响[6, 67]。与海水Mg/Ca和大气二氧化碳分压相比,海水碳酸盐饱和度属于次一级影响鲕粒原生矿物成分的因素。由于海水Mg/Ca并不能显著影响文石的沉淀速率,方解石和文石矿物溶度系数的差异造成两者在饱和度上也存在差异。
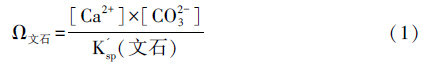
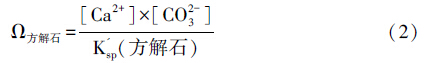
则Ω文石/Ω方解石=K'sp(方解石)/K'sp(文石)=4.39/6.65≈1/1.51 式中,K'sp(文石)=6.65×10-7 mol2 kg-2;K'sp(方解石)=4.39×10-7 mol2 kg-2 (K'sp为溶度积常数;引自[68])
当Ω文石=1X时,Ω方解石=1.5X;
当Ω方解石=1X时,Ω文石≈0.7X;
所以0.7X<Ω文石<1X时,1X< Ω方解石<1.5X。
方解石在海水中的饱和度(Ω方解石)比文石在海水中的饱和度(Ω文石)高1.5倍左右。例如,现代海水中方解石5倍超饱和,而文石是3.5倍。但是针状的文石晶体具有延伸的C轴,其生长速度要快于菱面体的方解石晶体,在同处于超饱和海水环境下由于晶体结构的差异,文石具有更快的生长速率;当海水饱和度降低,达到1X< Ω方解石<1.5X时,方解石仍处于超饱和状态而文石不饱和(0.7X<Ω方解石<1X)。这时海水中只能沉淀出方解石质的矿物,需要注意的是这种情况需要极高的(2 600~3 500 ppm)大气二氧化碳分压[67];当Ω方解石<1X时,文石和方解石均处于不饱和状态,不再形成碳酸盐矿物。
显生宙绝大多数时间内,海水中文石和方解石均处于稳定的超饱和状态。同时,长时间尺度下海水中镁离子和钙离子的输入、输出速率非常稳定,且混合速率非常快,整个海水中具有相对一致的Mg/Ca,并且Mg/Ca也不太可能在短时间尺度内(小于10 Ma)发生迅速变化。当洋中脊活动速率不发生大的变化时,一些极端事件可能会扰动(区域)海水化学成分,造成鲕粒原生矿物和分布情况的转变,比如:极端气候因素影响下温度、盐度以及碳酸盐饱和度的变化[6, 74];强烈的火山活动,造成短期大气二氧化碳分压的变化,促成海洋酸化及之后的陆缘风化反馈致使海水碳酸盐饱和度发生波动[75, 76]。但是,这种受约束的海水饱和度条件持续时间较短,在大气—水—岩石圈的调节作用下,海水化学成分能够及时进行调整。因此,极端事件对鲕粒原生矿物成分的影响可能只会出现在区域范围或短时间尺度下[77]。在更长的时间尺度下(大于10 Ma),海水Mg/Ca或是更为综合的因素(如温度,海水硫酸盐浓度,碱度等与Mg/Ca的协同变化)制约着碳酸盐鲕粒的矿物组成,对于这些问题还需要我们进行更为深入地研究。
4 结论及意义(1) 鲕粒原生矿物包括文石、低镁方解石和高镁方解石三种形式,既可以是单矿物组成,也可以由两种矿物共同组成。古代文石质鲕粒以切线状纹层结构为主,一般保存较差。部分文石鲕残存特殊的纹层结构(包括砖砌结构、溶解变形结构以及偏心结构等)。在封闭成岩环境下保存的文石质鲕粒往往具有较高的Sr浓度;高镁方解石鲕粒大多保留有较好的纹层结构,具有低Sr(一般<1 000 ppm)高Mg(4~20 mol % MgCO3),且具放射状或切线—放射状纹层结构。纹层由微亮晶方解石组成,部分纹层中含有具标志意义的微白云石包裹体颗粒;低镁方解石鲕粒绝大部分保留有原生精细结构,且具有低Sr(一般<1 000 ppm)和低Mg(0~4 mol % MgCO3)浓度以及放射状或切线—放射状纹层结构。另外,双矿物鲕粒可以根据两类纹层在结构和保存特征上的差异进行区分;
(2) 鲕粒原生纹层结构不只是简单地反应形成环境、水动力条件和盐度等信息,还蕴藏着指示古海洋化学特征的线索。鲕粒矿物成分的变化受到海水化学条件的控制,尤其是受到Mg/Ca,大气pCO2以及碳酸盐饱和度因素的影响。Mg/Ca在长时间尺度下(大于10 Ma)控制着鲕粒原生矿物类型;在短时间尺度(小于10 Ma)或区域范围内,Mg/Ca保持基本稳定的前提下,碳酸盐饱和度的变化可能对鲕粒矿物组成起着关键作用。
虽然鲕粒产出地层时代有限且在碳酸盐沉积记录中所占的比重较小,但是在一些关键地层中似乎“从未缺席”,它能够敏锐地捕捉到海水化学成分发生变化的信号。通过碳酸盐鲕粒原生矿物分析海水化学成分,是独立于古生物和地球化学分析之外的一种较为可靠的方法,这为研究古海洋化学特征提供了另一种途径。
致谢 感谢两位评审专家的宝贵意见。
| [1] | Sorby H C. The structure and origin of limestones[J]. Proceedings of the Geological Society of London, 1879, 35:56-95. |
| [2] | Richter D K. Classification of coated grains:discussion[M]//Peryt T M. Coated Grains. Berlin, Heidelberg:Springer-Verlag, 1983:7-8. |
| [3] | Flügel E. Microfacies of Carbonate Rocks:Analysis, Interpretation and Application[M]. Berlin, Heidelberg:Springer-Verlag, 2004:1-976. |
| [4] | Tucker M E, Wright V P. Carbonate Sedimentology[M]. USA:Wiley-Blackwell, 1990:1-496. |
| [5] | Opdyke B N, Wilkinson B H. Paleolatitude distribution of Phanerozoic marine ooids and cements[J]. Palaeogeography, Palaeoclimatology, Palaeoecology, 1990, 78(1/2):135-148. |
| [6] | Heydari E, Moore C H. Paleoceanographic and paleoclimatic controls on ooid mineralogy of the Smackover Formation, Mississippi salt basin-implications for Late Jurassic seawater composition[J]. Journal of Sedimentary Research, 1994, 64(1):101-114. |
| [7] | Ma Y S, Guo X S, Guo T L, et al. The Puguang gas field:new giant discovery in the mature Sichuan Basin, southwest China[J]. AAPG Bulletin, 2007, 91(5):627-643. |
| [8] | Duguid S M A, Kyser T K, James N P, et al. Microbes and ooids[J]. Journal of Sedimentary Research, 2010, 80(3):236-251. |
| [9] | Gaffey S J. Formation and infilling of pits in marine ooid surfaces[J]. Journal of Sedimentary Petrology, 1983, 53(1):193-208. |
| [10] | Folk R L, Lynch F L. Organic matter, putative nannobacteria and the formation of ooids and hardgrounds[J]. Sedimentology, 2001, 48(2):215-229. |
| [11] | Wright D T, Oren A. Nonphotosynthetic bacteria and the formation of carbonates and evaporites through time[J]. Geomicrobiology Journal, 2005, 22(1/2):27-53. |
| [12] | Plee K, Ariztegui D, Martini R, et al. Unravelling the microbial role in ooid formation-results of an in situ experiment in modern freshwater Lake Geneva in Switzerland[J]. Geobiology, 2008, 6(4):341-350. |
| [13] | Pacton M, Ariztegui D, Wacey D, et al. Going nano:a new step toward understanding the processes governing freshwater ooid formation[J]. Geology, 2012, 40(6):547-550. |
| [14] | Summons R E, Bird L R, Gillespie A L, et al. Lipid biomarkers in ooids from different locations and ages:evidence for a common bacterial flora[J]. Geobiology, 2013, 11(5):420-436. |
| [15] | 梅冥相. 鲕粒成因研究的新进展[J]. 沉积学报, 2012, 30(1):20-32. [Mei Mingxiang. Brief introduction on new advances on the origin of ooids[J]. Acta Sedimentologica Sinica, 2012, 30(1):20-32]. |
| [16] | Eardley A J. Sediments of Great Salt Lake, Utah[J]. AAPG Bulletin, 1938, 22(10):1305-1411. |
| [17] | Carozzi A V. Cerebroid oolites[J]. Transactions of the Illinois State Academy of Science, 1962, 55(3/4):238-249. |
| [18] | Shearman D J, Twyman J, Zand Karimi M. The genesis and diagenesis of oolites[J]. Proceedings of the Geologists' Association, 1970, 81(3):561-575. |
| [19] | Kahle C F. Ooids from Great Salt Lake, Utah, as an analogue for the genesis and diagenesis of ooids in marine limestones[J]. Journal of Sedimentary Research, 1974, 44(1):30-39. |
| [20] | Sandberg P A. New interpretations of Great Salt Lake ooids and of ancient non-skeletal carbonate mineralogy[J]. Sedimentology, 1975, 22(4):497-537. |
| [21] | Simone L. Ooids:a review[J]. Earth-Science Reviews, 1980, 16:319-355. |
| [22] | Marshall J F, Davies P J. High-magnesium calcite ooids from the Great Barrier Reef[J]. Journal of Sedimentary Petrology, 1975, 45(1):285-291. |
| [23] | Milliman J D, Barretto H T. Relict magnesian calcite oolite and subsidence of the Amazon shelf[J]. Sedimentology, 1975, 22(1):137-145. |
| [24] | Davies P J, Martin K. Radial aragonite ooids, Lizard Island, Great Barrier Reef, Queensland, Australia[J]. Geology, 1976, 4(2):120-122. |
| [25] | Halley R B. Ooid fabric and fracture in the Great Salt Lake and the geologic record[J]. Journal of Sedimentary Research, 1977, 47(3):1099-1120. |
| [26] | Medwedeff D A, Wilkinson B H. Cortical Fabrics in Calcite and Aragonite Ooids[M]//Peryt T M. Coated Grains.Berlin Heidelberg:Springer, 1983:109-115. |
| [27] | Heller P L, Komar P D, Pevear D R. Transport processes in ooid genesis[J]. Journal of Sedimentary Research, 1980, 50(3):943-951. |
| [28] | Wilkinson B H, Landing E. ""Eggshell diagenesis"" and primary radial fabric in calcite ooids[J]. Journal of Sedimentary Research, 1978, 48(4):1129-1138. |
| [29] | Deelman J C. Experimental ooids and grapestones:carbonate aggregates and their origin[J]. Journal of Sedimentary Research, 1978, 48(2):503-512. |
| [30] | Chow N, James N P. Facies-specific, calcitic and bimineralic ooids from Middle and Upper Cambrian platform carbonates, western Newfoundland, Canada[J]. Journal of Sedimentary Research, 1987, 57(5):907-921. |
| [31] | Friedman G M, Amiel A J, Braun M, et al. Generation of carbonate particles and laminites in algal mats:example from sea-marginal hypersaline pool, Gulf of Aqaba, Red Sea[J]. AAPG Bulletin, 1973, 57(3):541-557. |
| [32] | Strasser A. Ooids in Purbeck limestones (lowermost Cretaceous) of the Swiss and French Jura[J]. Sedimentology, 1986, 33(5):711-727. |
| [33] | Berner R A. The role of magnesium in the crystal growth of calcite and aragonite from sea water[J]. Geochimica Et Cosmochimica Acta, 1975, 39(4):489-494, IN3, 495-504. |
| [34] | Sandberg P A. An oscillating trend in Phanerozoic non-skeletal carbonate mineralogy[J]. Nature, 1983, 305(5929):19-22. |
| [35] | Hardie L A. Secular variation in seawater chemistry:an explanation for the coupled secular variation in the mineralogies of marine limestones and potash evaporites over the past 600 m. y.[J]. Geology, 1996, 24(3):279-283. |
| [36] | Lowenstein T K, Hardie L A, Timofeeff M N, et al. Secular variation in seawater chemistry and the origin of calcium chloride basinal brines[J]. Geology, 2003, 31(10):857-860. |
| [37] | Berner R A, Kothavala Z. Geocarb III:A revised model of atmospheric CO2 over Phanerozoic time[J]. American Journal of Science, 2001, 301(2):182-204. |
| [38] | Bathurst R G C. Carbonate Sediments and Their Diagenesis[M]. Amsterdam:Elsevier Science, 1972:1-620. |
| [39] | Loreau J P, Purser B H. Distribution and Ultrastructure of Holocene Ooids in the Persian Gulf [M]//Purser B H. The Persian Gulf. New York:Springer-Verlag, 1973:279-328. |
| [40] | Scholle P A, Kinsman D J. Aragonitic and high-Mg calcite caliche from the Persian Gulf-a modern analog for the Permian of Texas and New Mexico[J]. Journal of Sedimentary Research, 1974, 44(3):904-916. |
| [41] | Lippmann F. Sedimentary Carbonate Minerals[M]. New York:Springer-Verlag, 1973:1-228. |
| [42] | Wilkinson B H, Owen R M, Carroll A R. Submarine hydrothermal weathering, global eustasy, and carbonate polymorphism in Phanerozoic marine oolites[J]. Journal of Sedimentary Research, 1985, 55(2):171-183. |
| [43] | Algeo T J, Watson B A. Calcite, aragonite, and bimineralic ooids in the Missourian (Upper Pennsylvanian) strata of Kansas:Stratigraphic and geographic patterns of variation[M]//Pausé P H, Candelaria M P. Carbonate Facies and Sequence Stratigraphy:Practical Applications of Carbonate Models. USA:PBGC-SEPM Publication, 1995:141-173. |
| [44] | Major R P, Halley R B, Lukas K J. Cathodoluminescent bimineralic ooids from the Pleistocene of the Florida continental shelf[J]. Sedimentology, 1988, 35(5):843-855. |
| [45] | Li F, Yan J X, Algeo T, et al. Paleoceanographic conditions following the end-Permian mass extinction recorded by giant ooids (Moyang, South China)[J]. Global and Planetary Change, 2013, 105:102-120. |
| [46] | Lehrmann D J, Minzoni M, Li X W, et al. Lower Triassic oolites of the Nanpanjiang Basin, south China:facies architecture, giant ooids, and diagenesis—implications for hydrocarbon reservoirs[J]. AAPG Bulletin, 2012, 96(8):1389-1414. |
| [47] | Land L S, Behrens E W, Frishman S A. The ooids of Baffin Bay, Texas[J]. Journal of Sedimentary Research, 1979, 49(4):1269-1277. |
| [48] | Popp B N, Wilkinson B H. Holocene lacustrine ooids from Pyramid Lake, Nevada[M]//Peryt T M. Coated Grains. Berlin, Heidelberg:Springer-Verlag, 1983:142-153. |
| [49] | Rankey E C, Reeder S L. Holocene ooids of Aitutaki Atoll, Cook Islands, South Pacific[J]. Geology, 2009, 37(11):971-974. |
| [50] | Assereto R, Folk R L. Brick-like texture and radial rays in Triassic pisolites of Lombardy, Italy:a clue to distinguish ancient aragonitic pisolites[J]. Sedimentary Geology, 1976, 16(3):205-222. |
| [51] | Tucker M E. Calcitic, aragonitic and mixed calcitic-aragonitic ooids from the mid-Proterozoic Belt Supergroup, Montana[J]. Sedimentology, 1984, 31(5):627-644. |
| [52] | Tucker M E. Calcitized aragonite ooids and cements from the Late Precambrian Biri Formation of southern Norway[J]. Sedimentary Geology, 1985, 43(1/2/3/4):67-84. |
| [53] | Tewari V C, Tucker M E. Ediacaran Krol carbonates of the lesser Himalaya, India:stromatolitic facies, depositional environment and diagenesis[M]//Tewari V, Seckbach J. STROMATOLITES:Interaction of Microbes with Sediments. Netherlands:Springer, 2011, 18:133-156. |
| [54] | Wilkinson B H, Buczynski C, Owen R M. Chemical control of carbonate phases; implications from Upper Pennsylvanian calcite-aragonite ooids of southeastern Kansas[J]. Journal of Sedimentary Research, 1984, 54(3):932-947. |
| [55] | Richter D K. Calcareous ooids:a synopsis[M]//Peryt T M. Coated Grains. Berlin, Heidelberg:Springer-Verlag, 1983:71-99. |
| [56] | Brand U, Veizer J. Origin of coated grains:trace element constraints[M]//Peryt T M. Coated Grains. Berlin, Heidelberg:Springer-Verlag, 1983:9-26. |
| [57] | Lohmann K C, Meyers W J. Microdolomite inclusions in cloudy prismatic calcites:a proposed criterion for former high-magnesium calcites[J]. Journal of Sedimentary Research, 1977, 47(3):1078-1088. |
| [58] | Richter K D, Füchtbauer H. Ferroan calcite replacement indicates former magnesian calcite skeletons[J]. Sedimentology, 1978, 25(6):843-860. |
| [59] | Swirydczuk K. Mineralogical control on porosity type in Upper Jurassic Smackover ooid grainstones, southern Arkansas and northern Louisiana[J]. Journal of Sedimentary Research, 1988, 58(2):339-347. |
| [60] | Kinsman D J J. Interpretation of Sr2+ concentrations in carbonate minerals and rocks[J]. Journal of Sedimentary Petrology, 1969, 39(2):486-508. |
| [61] | Heydari E, Snelling R D, Dawson W C, et al. Ooid Mineralogy and Diagenesis of the Pitkin Formation, North-Central Arkansas[M]//Keith B D, Zuppann C W. AAPG Studies in Geology. Mississippian oolites and modern analogs. American Association of Petroleum Geologists, 1993:175-184. |
| [62] | Kidder D L, Hall S. Petrology and diagenetic evolution of Neoproterozoic ooids (Libby Formation, western Montana, U. S. A.)[J]. Precambrian Research, 1993, 63(1/2):83-96. |
| [63] | Singh U. Ooids and cements from the Late Precambrian of the Flinders Ranges, South Australia[J]. Journal of Sedimentary Petrology, 1987, 57(1):117-127. |
| [64] | Oomori T, Kaneshima H, Maezato Y. Distribution coefficient of Mg2+ ions between calcite and solution at 10-50℃[J]. Marine Chemistry, 1987, 20(4):327-336. |
| [65] | Swett K, Knoll A H. Marine pisolites from Upper Proterozoic carbonates of East Greenland and Spitsbergen[J]. Sedimentology, 1989, 36(1):75-93. |
| [66] | Mackenzie F T, Pigott J D. Tectonic controls of Phanerozoic sedimentary rock cycling[J]. Journal of the Geological Society, 1981, 138(2):183-196. |
| [67] | Morse J W, Wang Q W, Tsio M Y. Influences of temperature and Mg:Ca ratio on CaCO3 precipitates from seawater[J]. Geology, 1997, 25(1):85-87. |
| [68] | Morse J W, Arvidson R S, Lüttge A. Calcium carbonate formation and dissolution[J]. Chemical Reviews, 2007, 107(2):342-381. |
| [69] | Stanley S M, Hardie L A. Secular oscillations in the carbonate mineralogy of reef-building and sediment-producing organisms driven by tectonically forced shifts in seawater chemistry[J]. Palaeogeography, Palaeoclimatology, Palaeoecology, 1998, 144(1/2):3-19. |
| [70] | Ries J B. Review:geological and experimental evidence for secular variation in seawater Mg/Ca (calcite-aragonite seas) and its effects on marine biological calcification[J]. Biogeosciences, 2010, 7(9):2795-2849. |
| [71] | Stanley S M, Hardie L A. Hypercalcification:paleontology links plate tectonics and geochemistry to sedimentology[J]. GSA Today, 1999, 9(2):1-7. |
| [72] | Lowenstein T K, Timofeeff M N, Brennan S T, et al. Oscillations in Phanerozoic seawater chemistry:evidence from fluid inclusions[J]. Science, 2001, 294(5544):1086-1088. |
| [73] | Kiessling W, Aberhan M, Villier L. Phanerozoic trends in skeletal mineralogy driven by mass extinctions[J]. Nature Geoscience, 2008, 1(8):527-530. |
| [74] | Liu J B, Zhan R B, Dai X, et al. Demise of Early Ordovician oolites in South China:Evidence for paleoceanographic changes before the GOBE[M]//Gutiérrez-Marco J C, Rábano I, García-Bellido D. Ordovician of the World. Cuadernos del Museo Geominero, 2011:309-317. |
| [75] | Payne J L, Turchyn A V, Paytan A, et al. Calcium isotope constraints on the end-Permian mass extinction[J]. Proceedings of the National Academy of Sciences of the United States of America, 2010, 107(19):8543-8548. |
| [76] | Hönisch B, Ridgwell A, Schmidt D N, et al. The geological record of ocean acidification[J]. Science, 2012, 335(6072):1058-1063. |
| [77] | Li F, Yan J X, Chen Z Q, et al. Global oolite deposits across the Permian-Triassic boundary:A synthesis and implications for palaeoceanography immediately after the end-Permian biocrisis[J]. Earth-Science Reviews, 2015, doi:10.1016/j.earscirev.2014,12.006. |