 2017, Vol. 36
2017, Vol. 36



锰元素(Mn)在地球中的含量丰富(Turekian and Wedepohl, 1961),易被氧化形成锰氧化物矿物,在陆相沉积物中广泛分布(Post,1992)。在近地球表面,已知的锰氧化物/水合锰氧化物矿物达30多种,在金属和有机碳生物地球化学中扮演着重要角色(Post,1999)。
自Morita(1977)及Trasatti(1980)等提出MnO2是优良的产氧光催化剂后,Cao和Suib(1994)首次通过光催化氧化2-丙醇证明无论是晶体锰氧化物还是无定形的锰氧化物都具有光催化氧化活性。锰氧化物矿物作为半导体材料成为研究的热点和前沿。Sherman(2005)通过化学法合成水钠锰矿,研究了其在水溶液环境中光催化自还原溶解的热力学性质。Pinaud等(2011)通过电沉积薄膜法合成水钠锰矿晶体,研究其光学特性和太阳能光电化学性质。前人对于锰氧化物矿物光催化氧化还原水的方法和技术也展开了大量的研究(Armaroli and Balzani, 2006;Dasgupta et al., 2010;Robinson et al., 2013)。同时,由于纳米材料具有的表面效应、小尺寸效应和宏观量子隧穿效应等特点,研究合成低维纳米结构锰氧化物半导体材料也吸引着越来越多的目光(Ahmed and Huang, 2012)。
虽然锰氧化物矿物光催化研究受到普遍关注,但对于锰氧化物矿物能带结构理论的分析鲜有研究。在光催化反应中,半导体能带结构决定光生电子和空穴产生和被激发的条件,以及激发后与电解质溶液中氧化还原电对的相互作用过程,所以,了解半导体能带结构对于光催化研究十分重要。同时,地表锰氧化物和有机物尤其是腐殖质之间发生的氧化还原反映,锰氧化物光催化腐殖质的氧化分解过程与机理还有待进一步研究。本文通过合成碱性水钠锰矿、酸性水钠锰矿、锰钾矿、δ-MnO2几种不同结构类型的锰氧化物矿物,以XRD、ICP、ZETA电势仪、Raman、同步辐射等测试手段,对其结构、组成和化学性质进行表征,通过理论计算,初步探讨了pH条件下物相及结构类型对能带位置的影响,并对普遍存在于沉积物和土壤中的锰氧化物矿物与腐殖质光催化氧化还原电势之间的关系进行了详细研究。
1 材料与方法 1.1 样品合成碱性水钠锰矿采用改进的Giovanoli法,在低温碱性介质中氧化MnCl2合成(Giovanoli et al., 1973;冯雄汉, 2003, 冯雄汉等,2003):将冷却至10℃以下的250 mL 10 mol/L的KOH溶液迅速倒入250 mL 0.8 mol/L的MnSO4溶液中混合曝气,能气量为30 L/min,冰水浴控制实验反应温度低于5℃,开启磁力搅拌450 r/min,反应5 h。
酸性水钠锰矿按照OPP方法在酸性介质中还原KMnO4合成(冯雄汉,2003):将300 mL浓度为0.4 mol/L的KMnO4溶液用加热磁力搅拌器以450 r/min转速加热至沸腾,按0.7 L/min的速率滴加35 mL浓度为12 mol/L的浓盐酸,滴加完毕后在沸腾条件反应30 min。
锰钾矿根据改进的Mckenzie法,在CH3COOH溶液中氧化还原MnSO4、KMnO4合成(Mckenzie,1971;冯雄汉,2003):取0.05 mol MnSO4加入到100 mL浓度为2 mol/L的乙酸溶液中,加热至60℃后,与60℃的80 mL 0.44 mol/L的KMnO4溶液混合,置于加热磁力搅拌器上,以450 r/min转速加热至沸腾,回流恒温搅拌1 h。
δ-MnO2在常温弱碱条件下按照Balistrieri和Murray(1982)方法合成:用100 mL浓度为0.139 mol/L的KMnO4与0.286 mol/L的KOH混合溶液,按10 mL/min的速率滴定1 L浓度为0.056 mol/L的Mn(NO3)2,置于磁力搅拌器上,以450 r/min转速搅拌反应1 h。
实验所用试剂均为分析纯,所用水为去离子水(DDW,电阻率18 MΩ),反应结束后,将生成的矿物洗涤、干燥保存。
1.2 样品表征 1.2.1 粉晶X射线衍射(XRD)将样品研磨过200目筛,压片,用D/max-rA型粉晶衍射仪(Rigaku公司,日本)对样品进行物相鉴定。测试条件为Cu Kα辐射,管压40 kV,管流100 mA,扫描速度8(°)/min,步长0.02°扫描范围5°~80°。
1.2.2 Raman散射粉末样品,用Renishaw Invia Reflex拉曼光谱仪测定。测试条件为扫描范围0~4000 cm-1,激发波长532 nm,累计扫描次数40次。
1.2.3 ICP和AOS测定Mn、Na和K元素含量通过ICP-OES仪器(PECTRO BLUE SOP)进行分析。测试条件:功率1.00 kW,载气为氩气,等离子气体15.0 L/min,辅助气1.5 L/min,雾化气200 kPa,泵速15 r/min,清洗时间10 s,重复读数3次,自动调节积分时间。
锰平均氧化度(AOS)采用高锰酸钾反滴定法测定(Kijima et al., 2001):将5.0 mL H2C2O4溶液(0.5 mol/L)、10.0 mL H2SO4溶液(0.5 mol/L)混合,置于磁力搅拌器上加热至60℃,称取0.1000 g样品加入混合溶液中。加入样品后开始搅拌并用0.02 mol/L KMnO4溶液滴定至反应结束,搅拌速率450 r/min。待溶溶液由无色变为浅粉紫色,后迅速变为亮黄棕色。保持30 s不褪色即认为达到终点,记录高锰酸钾滴定体积。每个样品重复测定3次,取均值。
合成锰氧化物矿物中锰的平均价态(AOS)可通过下式计算(Kijima et al., 2001)。
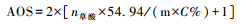
|
(1) |
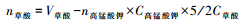
|
(2) |
式中:n草酸为草酸反应体积(mL);n高锰酸钾为高锰酸钾滴定体积(mL);m为样品质量(g);C%为样品中总Mn质量分数(通过ICP测试,依据化学组成换算);V草酸为反应体系中加入草酸的总体积;C高锰酸钾为高锰酸钾浓度;C草酸为草酸浓度;
1.2.4 Zeta电势测定样品表面零电荷点(PZC)采用Zeta电势法(Kosmulski,2002),通过Zeta电势仪(Brookhaven-NanoBrook Omni)测定:用1 mmol/L NaCl(KCl)为电解质溶液配制一系列0.01 g/L的样品溶液,以HCl或NaOH(KOH)溶液调节成梯度pH的溶液,静置2~3天。用Zeta电势仪测定样品在不同pH值下的Zeta电势。通过Zeta电势对pH作图,当Zeta电势为零时对应的pH即为样品的零电点。
1.2.5 同步辐射测定禁带宽度锰氧化物的禁带宽度通过同步辐射氧K边吸收发射光谱测得,氧K边吸收谱及发射谱是在加拿大光源(Canadian Light Source,CLS)的10ID-2线站测得。狭缝宽为25 mm。吸收谱分辨率为0.4 eV,发射谱分辨率0.6 eV。将样品用酒精分散滴于碳胶上,待干后放入样品腔中,实验在真空度小于10-9的环境下进行。吸收谱的测定模式为全电子产额(total electron yield,TEY)。
2 结果与分析 2.1 矿物物相分析图 1为实验所用合成的几种锰氧化物矿物样品的粉晶X射线衍射图谱。根据图 1可知,合成碱性水钠锰矿XRD图谱的3个强峰对应的2θ角为12.4°、24.9°,与PDF#43-1456卡片一致,分别对应的晶面为(001)、(002),说明合成的碱性水钠锰矿为单斜水钠锰矿。根据图 1可知,合成的酸性水钠锰矿XRD图谱三强峰对应的2θ角为12.3°、24.8°、37.6°,分别对应的晶面为(001)、(002)、(012),与PDF#86-0666一致,说明合成的酸性水钠锰矿为六方水钠锰矿。δ-MnO2属于水钠锰矿族矿物,其XRD

|
图 1 合成氧化锰矿物XRD图谱 Figure 1 XRD pattern of synthetic Mn oxide minerals |
图谱与水钠锰矿相比,(001) 和(002) 晶面对应的谱峰缺失或很弱(Chukhrov et al., 1987)。由合成样品XRD图谱(图 1)可知,样品的(001) 和(002) 晶面缺失,其特征峰对应2θ角为37.6°、65.6°,与PDF#15-0604匹配,因而合成样品为δ-MnO2。将合成锰钾矿样品的XRD图谱(图 1)与PDF#12-0706对比,样品(110)、(310)、(211) 晶面对应3个强峰的2θ角为12.8°、28.5°、37.4°,与PDF#12-0706吻合,可知合成锰钾矿为四方晶系锰钾矿。
实验用的几种合成锰氧化物矿物样品的Raman光谱如图 2所示。Raman光谱对难以应用XRD数据进行精细分析的短程有序非晶体化合物和结晶度差的矿物非常敏感(Julien et al., 2002),结合Raman与XRD图谱对合成矿物进行表征可以对锰氧化物矿物的矿物学特征进行进一步分析。

|
图 2 锰氧化物矿物拉曼光谱 Figure 2 Raman spectra of Mn oxide minerals |
合成锰钾矿拉曼光谱(图 2)显示有3个强峰:181 cm-1、572 cm-1、631 cm-1,4个弱峰:351 cm-1、384 cm-1、479 cm-1、514 cm-1。应用因子群论对属于I/4 m空间群的锰钾矿振动类型进行分析,因为锰钾矿是含有掺杂离子的2×2隧道结构,所以Mn-O振动和锰氧八面体形成的双链振动所对应的拉曼活性是锰钾矿固有的属性,572 cm-1、631 cm-1处对应的尖锐的强峰说明样品为2×2隧道结构,四方晶系,结晶度好(Gao et al., 2008)。此外,由于锰钾矿掺杂的隧道离子占据2a和2b位点,在其对应的C4 h点群中并不产生Raman活性(Fateley et al., 1971)。据前人研究(Julien et al., 2002;Ma et al., 2005),183 cm-1处低频拉曼谱带归属于锰氧八面体平移产生的面外弯曲振动。
与锰钾矿对比分析碱性水钠锰矿、酸性水钠锰矿、δ-MnO2的拉曼光谱,尽管4个样品在570 cm-1、630 cm-1附近具有锰氧八面体以及Mn-O特征振动,但是由于层间离子及构造类型的不同导致拉曼特征谱带有细微差异性(Julien et al., 2004)。Julien等(2003)研究表明:碱性水钠锰矿、酸性水钠锰矿、δ-MnO2等水钠锰矿族锰氧化物矿物的特征峰主要分布在500~510 cm-1、570~585 cm-1和625~650 cm-1区域,625~650 cm-1是在锰氧八面体群的υ2(Mn-O)对称伸缩振动;而570~585 cm-1是在锰氧八面体层基面的υ3(Mn-O)伸缩振动,对层状水钠锰矿族矿物尤其强烈,是水钠锰矿族矿物的指纹特征区。
结合XRD和Raman谱图分析结果,可以确定合成的4种样品分别为碱性水钠锰矿、酸性水钠锰矿、锰钾矿、δ-MnO2。
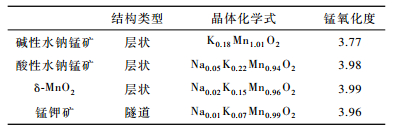
2.2 晶体化学式与PZC分析样品元素含量及锰氧化度列于表 1。通过对合成的4种锰氧化物矿物的Zeta电势-pH作图(图 3)分析得到其零电荷点。结果如图 3所示:碱性水钠锰矿、酸性水钠锰矿、δ-MnO2、锰钾矿的PZC分别为3.0、1.8、3.6、1.8。
|
|
表 1 锰氧化物矿物样品化学分析数据 Table 1 Chemical properties of Mn oxide minerals |

|
图 3 锰氧化物Zeta电位图 Figure 3 Zeta potential of Mn oxide minerals |
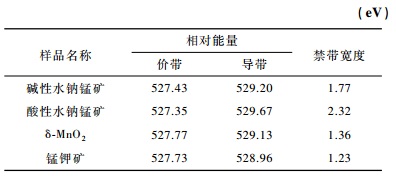
当半导体矿物吸收光电子后,电子由最高占有轨道(HOMO)跃迁到最低空轨道(LUMO),HOMO相互作用形成价带,LUMO相互作用形成导带,而禁带宽度为导带和价带之间的能量差。Sherman等(2005)通过氧K边X射线吸收及发射图谱谱峰半高宽所对应的导、价带能量,对针铁矿、赤铁矿、纤铁矿、软锰矿、锰钾矿及水钠锰矿等金属氧化物的禁带宽度进行研究计算。丁聪等(2016)利用同步辐射X射线氧的K边吸收谱与发射谱对纯针铁矿及天然针铁矿的能带结构进行了测定,与相关文献中实验结果相符。本文通过氧K边X射线吸收及发射图谱对锰氧化物禁带宽度进行研究计算,在锰氧化物晶体结构中,O 2p电子态与Mn 3d电子态发生重叠,Mn-O杂化成键,从而使得电子能够从O(1 s)轨道跃迁到未被占据的Mn(3d)轨道,产生吸收谱,当电子再回落到O(1 s)轨道的空穴时产生发射谱。吸收谱峰对应O(1 s)-Mn(3d),发射谱对应O(2p)-O(1 s)。图 4为合成的锰氧化物矿物的氧K边X射线吸收、发射图谱,样品的禁带宽度见表 2。
|
|
表 2 导带、价带相对能量与禁带宽度值 Table 2 Relative energies of valence and conduction bands |

|
图 4 合成锰氧化物矿物氧K边X射线吸收谱和发射谱 Figure 4 Oxygen K-dge XAS and XES of synthetic Mn oxide minerals |
大多数纯半导体材料中,价带对应电子的最高占有轨道,导带对应电子的最低空轨道。因此,Sherman(2005)提出:价带能级位置EVB可由化合物电离电势测量,导带能级位置ECB可由化合物电子亲和能测量,费米能级EF则是半导体化合物的绝对电负性。而天然半导体矿物半导体结构中都含有杂质,会导致禁带存在电子受体能级或供体能级,使半导体矿物的费米能级发生改变(p-型半导体EF稍高于EVB,n-型半导体EF略低于ECB)(Morrison,1990)。而大部分金属氧化物半导体材料的费米能级与pH具有线性关系,称为能斯特线性关系(Butler and Ginley, 1978;Halouani and Deschanvres, 1982;Matsumoto et al., 1989)。同时,Butler和Ginley(1978)首次提出半导体/水溶液体系中能级位置可以通过电负性预测。而元素电负性是Huheey(1972)通过对元素的电离能和电子亲和能计算得出的。Xu和Schoonen(2000)认为化合物的电负性为元素几何均值,并在前人研究基础上提出化合物AVS模式下能级计算的理论经验公式:
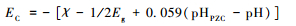
|
(3) |
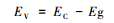
|
(4) |
式中:χ为化合物几何平均电负性,Eg为禁带宽度;pHPZC为零电点。而AVS与氧化还原电势对应的标准氢电极电势转换关系如下式:
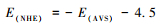
|
(5) |
AxByCz化合物的几何平均电负性计算公式为:
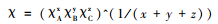
|
(6) |
式中:χA、χB、χC为元素A、B、C的密立根电负性(Huheey,1972)。
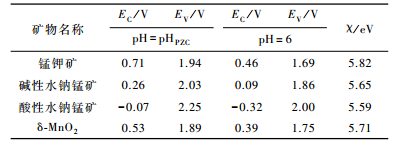
根据经验公式和实验测得的禁带宽度、PZC、χ(见表 2),可计算锰氧化物矿物的导带、价带能级位置。表 3为pH=pHPZC和pH=6时样品导带、价带的能级位置。
|
|
表 3 不同pH条件下导/价带能级位置与绝对电负性 Table 3 The energies of valence and conduction band at different pH condition and absolute electronegativity |
由于合成的锰氧化物矿物的禁带宽度能够在可见光下激发半导体产生载流子,在O(2p)和Mn(3d)杂化轨道产生空穴,Mn(3d)轨道生成电子,光生空穴及电子可与环境介质中的有机物和无机物发生氧化还原反应(Sherman,2005)。Sunda和Kieber(1994)提出锰氧化物可以将不能直接被微生物利用的腐殖质降解为可促进生物生长的小分子化合物,并通过实验证明锰氧化物可以与腐殖质反应生成小分子有机物。
Scott等(1998)认为腐殖质的半醌自由基是微生物与环境介质反应电子传输的载体。Österberg和Shirshova(1997)在测量腐殖质氧化还原电动电势时认为腐殖质电动电势与溶液pH之间也符合能斯特方程线性关系。因而,在前人研究腐殖质电动电势和锰氧化物能带位置与pH相关性的基础上,结合半导体材料能级经验公式(3),笔者对不同pH条件下4种锰氧化物矿物的能级位置进行理论计算,并作图与HA氧化还原电势进行比较分析(图 5)(Struyk and Sposito, 2001)。

|
图 5 锰氧化物能级位置与HA氧化还原电势关系图 Figure 5 The relationship between energy edge of Mn oxides and redox potential of HA |
如图 5所示,4种锰氧化物矿物的导、价带电势随着pH的增大而降低,即随着pH的增加导带电子还原能力更强、价带空穴氧化能力减弱。在pH=PZC时,酸性水钠锰矿、碱性水钠锰、δ-MnO2、锰钾矿对应的导带电势分别为-0.07 V、0.26 V、0.53 V、0.71 V,价带电势为2.25 V、2.03 V、1.89 V、1.94 V。在pH=6时,酸性水钠锰矿、碱性水钠锰、δ-MnO2、锰钾矿对应的导带电势分别为-0.32 V、0.09 V、0.39 V、0.46 V,价带电势为2.00 V、1.86 V、1.75 V、1.69 V。在任何pH条件下合成的4种锰氧化物矿物导带电极电势:酸性水钠锰矿 < 碱性水钠锰矿 < δ-MnO2 < 锰钾矿,价带电势:酸性水钠锰矿>碱性水钠锰矿>δ-MnO2>锰钾矿。因此,样品的光催化氧化还原能力为:酸性水钠锰矿>碱性水钠锰矿>δ-MnO2>锰钾矿。与腐殖质氧化还原电势相比较,4种锰氧化物矿物在任何pH范围内均有比腐殖质电极电势更高的价带电势,所以在热力学上4种锰氧化物矿物均能吸收太阳光中的可见光催化氧化腐殖质。
样品的光催化氧化还原能力为:酸性水钠锰矿>碱性水钠锰矿>δ-MnO2>锰钾矿,同时,酸性水钠锰矿、碱性水钠锰、δ-MnO2、锰钾矿的禁带宽度分别为:2.32 eV、1.77 eV、1.36 eV、1.23 eV,从结构上来看:3种层状结构的锰氧化物矿物(酸性水钠锰矿、碱性水钠锰、δ-MnO2)比隧道结构的锰钾矿具有更大的禁带宽度和更强的氧化还原能力。所以吸附在合成的层状结构锰氧化物矿物表面的腐殖质更能有效的利用光生空穴和电子发生氧化还原反应。故在热力学上,合成的层状的锰氧化物矿物相比于隧道结构的锰钾矿利用可见光光催化氧化腐殖质的能力更强。
3 结论合成的锰氧化物矿物:酸性水钠锰矿、碱性水钠锰、δ-MnO2、锰钾矿禁带宽度小于3.1 eV,均能吸收可见光激发产生光电子和空穴。样品的光催化氧化还原能力为:酸性水钠锰矿>碱性水钠锰矿>δ-MnO2>锰钾矿,与腐殖质氧化还原电势相比较,合成的4种锰氧化物矿物在任何pH范围内均有比腐殖质电极电势更高的价带电势,所以在热力学上合成的4种锰氧化物矿物均能吸收太阳光中的可见光催化氧化腐殖质。层状结构的锰氧化物比隧道结构的锰氧化物可见光催化氧化还原能力更强。
| [] | Ahmed K A M, Huang K X. 2012. Synthesis, Characterization and catalytic activity of birnessite type potassium manganese oxide nanotubes and nanorods. Materials Chemistry and Physics, 133(2-3): 605–610. DOI:10.1016/j.matchemphys.2012.01.009 |
| [] | Armaroli N, Balzani V. 2006. The future of energy supply: Challenges and opportunities. Angewandte Chemie International Edition, 46(1-2): 52–66. |
| [] | Balistrieri L S, Murray J W. 1982. The surface chemistry of δMnO2 in major ion seawater. Geochim Cosmochim Aeta, 46(6): 1041–1052. DOI:10.1016/0016-7037(82)90057-6 |
| [] | Butler M A, Ginley D S. 1978. Temperature dependence of flatband potentials at semiconductor-electrolyte interfaces. Nature, 273(5663): 524–525. DOI:10.1038/273524a0 |
| [] | Cao H, Suib S L. 1994. Highly efficient heterogeneous photooxidation of 2-propanol to acetone with amorphous manganese oxide catalysts. Journal of the American Chemical Society, 116(12): 5334–5342. DOI:10.1021/ja00091a044 |
| [] | Chukhrov F V. 1980. On vernadite. International Geology Review, 22(1): 58–74. DOI:10.1080/00206818209466863 |
| [] | Dasgupta J, Tyryshkin A M, Baranov S V, Dismukes C G. 2010. Bicarbonate coordinates to Mn3+ during photo-assembly of the catalytic Mn4Ca core of photosynthetic water oxidation: EPR characterization. Applied Magnetic Resonance, 37: 137–150. DOI:10.1007/s00723-009-0053-z |
| [] | Fateley W G, Mcdevitt N T, Bentley F F. 1971. Infrared and Raman selection rules for lattice vibrations: The correlation method. Applied Spectroscopy, 25(2): 155–173. DOI:10.1366/000370271779948600 |
| [] | Gao T, Glerup M, Krumeich F, Nesper R, Fjellvåg H, Norby P. 2008. Microstructures and spectroscopic properties of cryptomelane-type manganese dioxide nanofibers. Journal of Physical Chemistry C, 112(34): 13134–13140. DOI:10.1021/jp804924f |
| [] | Giovanoli R, Bühler H, Sokolowska K. 1973. Synthetic lithiophorite: Electron microscopy and X-ray diffraction. Journal de Microscopie, 18: 271–284. |
| [] | Halouani F E, Deschanvres A. 1982. Interfaces semi-conducteur-electrolyte: Correlations entre le potentiel de bande plate et les echelles d'electronegativite. Materials Research Bulletin, 17(8): 1045–1052. DOI:10.1016/0025-5408(82)90130-1 |
| [] | Huheey J E. 1972. Inorganic chemistry: Principles of structure and reactivity.New York: Harpar & Row. |
| [] | Julien C M, Massot M, Poinsignon C. 2004. Lattice vibrations of manganese oxides: Part Ⅰ.Periodic structures. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 60(3): 689–700. DOI:10.1016/S1386-1425(03)00279-8 |
| [] | Julien C, Massot M, Baddour-Hadjean R, Franger S, Bach S, Pereira-Ramosd J P. 2003. Raman spectra of birnessite manganese dioxides. Solid State Ionics, 159(3-4): 345–356. DOI:10.1016/S0167-2738(03)00035-3 |
| [] | Julien C, Massot M, Rangan S, Lemal M, Guyomard D. 2002. Study of structural defects in γ-MnO2 by Raman spectroscopy. Journal of Raman Spectroscopy, 33(4): 223–228. DOI:10.1002/(ISSN)1097-4555 |
| [] | Kijima N, Yasuda H, Sato T, Yoshimura Y. 2001. Preparation and characterization of open tunnel oxide α-mno2 precipitated by ozone oxidation. Journal of Solid State Chemistry, 159(1): 94–102. DOI:10.1006/jssc.2001.9136 |
| [] | Kosmulski M. 2002. The pH-dependent surface charging and the points of zero charge. Journal of Colloid and Interface Science, 253(1): 77–87. DOI:10.1006/jcis.2002.8490 |
| [] | Ma S B, Lee Y H, Ahn K Y, Kim C M, Oh K H, Kim K B. 2005. Spontaneously deposited manganese oxide on acetylene black in an aqueous potassium permanganate solution. Journal of the Electrochemical Society, 153(1): C27–C32. |
| [] | Matsumoto Y, Yoshikawa T, Sato E. 1989. Dependence of the band bending of the oxide semiconductors on pH. Journal of Electrochemical Society, 136(5): 1389–1391. DOI:10.1149/1.2096927 |
| [] | Mckenzie R M. 1971. The synthesis of birnessite, cryptomelane, and some other oxides and hydroxides of manganese. Mineralogical Magazine, 38(296): 493–502. DOI:10.1180/minmag |
| [] | Morita M, Iwakura C, Tamura H. 1977. The anodic characteristics of manganese dioxide electrodes prepared by thermal decomposition of manganese nitrate. Electrochimica Acta, 22(4): 325–328. DOI:10.1016/0013-4686(77)85081-0 |
| [] | Morrison S R. 1990. The chemical physics of surfaces.New York: Springer: 438. |
| [] | Österberg R, Shirshova L. 1997. Oscillating, nonequilibrium redox properties of humic acids. Geochimica et Cosmochimica Acta, 61(21): 4599–4604. DOI:10.1016/S0016-7037(97)00266-4 |
| [] | Pinaud B A, Chen Z B, Abram D N, Jaramillo T F. 2011. Thin films of sodium birnessite-type MnO2: Optical properties, electronic band structure, and solar photoelectrochemistry. Journal of Physical Chemistry C, 115(23): 11830–11838. DOI:10.1021/jp200015p |
| [] | Post J E. 1992. Crystal structures of manganese oxide minerals. Catena Supplement, 21: 51–73. |
| [] | Post J E. 1999. Manganese oxide minerals: Crystal structures and economic and environmental significance. Proceedings of the National Academy of Sciences of the United States of America, 96(7): 3447–3454. DOI:10.1073/pnas.96.7.3447 |
| [] | Robinson D M, Go Y B, Mui M, Gardner G, Zhang Z J, Mastrogiovanni D, Garfunkel E, Li J, Greenblatt M, Dismukes G C. 2013. Photochemical water oxidation by crystalline polymorphs of manganese oxides: Structural requirements for catalysis. Journal of the American Chemical Society, 135(9): 3494–3501. DOI:10.1021/ja310286h |
| [] | Scott D T, Mcknight D M, Blunt-Harris E L, Kolesar S E, Lovley D R. 1998. Quinone moieties act as electron acceptors in the reduction of humic substances by humics-reducing microorganisms. Environ ̄mental Science & Technology, 32(19): 2984–2989. |
| [] | Sherman D M. 2005. Electronic structures of iron(Ⅲ)and manganese(Ⅳ)(hydr)oxide minerals: Thermodynamics of photochemical reductive dissolution in aquatic environments. Geochimica et Cosmochimica Acta, 69(13): 3249–3255. DOI:10.1016/j.gca.2005.01.023 |
| [] | Struyk Z, Sposito G. 2001. Redox properties of standard humic acids. Geoderma, 102(3-4): 329–346. DOI:10.1016/S0016-7061(01)00040-4 |
| [] | Sunda W G, Kieber D J. 1994. Oxidation of humic substances by manganese oxides yields low-molecular-weight organic substrates. Nature, 367(6458): 62–64. DOI:10.1038/367062a0 |
| [] | Trasatti S. 1980. Electrocatalysis by oxides-Attempt at a unifying approach. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry, 111(1): 125–131. DOI:10.1016/S0022-0728(80)80084-2 |
| [] | Turekian K K, Wedepohl K H. 1961. Distribution of the elements in some major units of the earth's crust. Geological Society of America Bulletin, 72(2): 175–192. DOI:10.1130/0016-7606(1961)72[175:DOTEIS]2.0.CO;2 |
| [] | 丁聪, 李艳, 李岩, 鲁安怀. 2016. 同步辐射软X射线吸收谱与发射谱测定天然针铁矿能带结构. 岩石矿物学杂志, 35(2): 349–354. |
| [] | 冯雄汉, 谭文峰, 刘凡, 许永胜. 2003. 碱性介质中水钠锰矿的合成与转化. 矿物岩石地球化学通报, 22(2): 184–187. |
| [] | 冯雄汉. 2003. 几种常见氧化锰矿物的合成、转化及表面化学性质.武汉: 华中农业大学. |