 2017, Vol. 36
2017, Vol. 36



2. 中国地质大学 地质过程与矿产资源国家重点实验室, 北京 100083;
3. 中国科学院大学, 北京 100049
2. State Key Laboratory of Geological Processes and Mineral Resources, China University of Geosciences(Beijing), Beijing 100083, China;
3. University of Chinese Academy of Sciences, Beijing 100049, China
硒(Se)是人与动物必需的微量元素。人体若长期硒摄入量低于0.1 mg/kg, 会引起缺硒性反应症;高于1 mg/kg的会引起硒中毒(Tan et al., 2002;Dumont et al., 2006;Zhu et al., 2008)。在近期的报道中, 高放废物地质处置中的核反应过程会产生长半衰期核素 79Se(t1/2=3.77×105 a)(Bienvenu et al., 2007)。79Se具有化学和辐射双重毒性, 是高放废物地质处置中重点关注的几个放射性核素之一(Chen et al., 1999;Grambow, 2008)。因此, 环境中硒的地球化学行为及生物可利用性一直备受环境、土壤、生命安全等科学领域的关注(Herring, 1991;Finkelman et al., 1999;Zhu et al., 2004, 2014;Fordyce, 2007;张莹等, 2007;Clark and Johnson, 2008;Winkel et al., 2012;Mitchell et al., 2013;Schilling et al., 2014)。
硒与硫是同族元素, 具有类似硫的地球化学性质。氧化条件下, 以Se(Ⅵ)(SeO42-等)和Se(Ⅳ)(SeO32-、HSeO3-等)的形态为主, 前者具有较高的可溶性、易迁移, 而Se(Ⅳ)更易吸附于铁锰氧化物、有机质、黏土矿物等的表面(Zhu et al., 2004)。在还原环境中, 主要以元素Se(0)或硒化物(-Ⅱ)形式存在(Zhu et al., 2012)。元素Se或硒化物广泛分布在岩石、矿体、沉积物、土壤中(Zhu et al., 2004;Winkel et al., 2012)。如在高放废物地质处置库周围环境, 常通过还原-沉淀作用以亚硒酸形式存在, 79Se转化为难溶于水的元素硒或硒化物(Chen et al., 1999)。在一些砂岩型铀矿中也已发现存在元素硒和硒化物组合(Thompson et al., 1956)。
已有研究表明, H2O2是As(Pettine et al., 1999)、Fe(González-Davila et al., 2005)等的重要氧化剂。元素Se是硒化物氧化的初级产物, 同时也是高价溶解态硒还原沉淀的主要产物(Kang et al., 2013;Ma et al., 2014), 但环境中H2O2与元素Se作用的动力学规律目前仍缺乏详实的研究报道。因此, 开展元素Se与H2O2作用的研究对理解环境中Se的氧化还原行为和潜在环境风险评估具有重要意义。为此, 本文主要采用氢化物发生器-原子荧光光谱法(Zhu et al., 2004;秦海波等, 2008;朱建明等, 2011; Winkel et al., 2012)(以下简称HG-AFS), 重点研究在一定条件下元素硒(Se0)被H2O2氧化至硒氧离子过程中的反应动力学及其机理。以期能够为后期元素硒氧化过程中的硒同位素分馏研究奠定基础, 也能够为环境中硒的扩散、迁移、污染等的控制提供一定的参考, 进而期望为地质早期海洋与大气氧的演化、地质环境中早期生命形成与演变的研究提供一定的证据。
1 材料与方法 1.1 实验材料与试剂本试验中使用的元素硒粉末购于上海山浦化工有限公司, 纯度优于99.9%。元素硒粉末呈现分散的单个微米级硒颗粒或硒颗粒的团聚体;亚硒酸钠固体(Na2SeO3)购自天津市光复精细化工研究所, 纯度优于97.0%;Se(Ⅳ)标准储备液(100 μg/mL)购自国家标准物质中心, 保存于4 ℃冷藏柜中。标准工作液由储备液使用稀盐酸溶液逐级稀释, 称量法配制, 保存于100 mL棕色容量瓶中;双氧水(优级纯)买自国药集团化学试剂有限公司, 优级纯级NaOH购于天津科密欧化学试剂有限公司;HNO3和HCl购买于Fisher公司, 为Trace metal级。
实验用水均使用18.2 MΩ的Milli-Q超纯水(简称MQ水)。为排除氧气干扰, 使用前均用高纯氮气鼓泡1 h, 最终获得零溶解氧的MQ水(zero dissolved oxygen water, 以下简称ZDO水)。100 mL 0.8 mol/L NaOH+3% H2O2的混合储备液使用ZDO水配制, 使用前高纯氮鼓气1 h;称取Na2SeO3固体79.6 mg, 配制成硒浓度为460 μg/mL的Na2SeO3储备液(中性), 并用0.22 μm的特氟龙无菌滤头过滤, 置于4℃冰箱保存备用。
AG1-X8阴离子树脂(200~400目)使用前均活化洗涤, 保存于0.1 mol/L HCl溶液中;Biorad柱使用前分别用MQ水和6 mol/L HCl浸泡洗涤;实验所使用的玻璃器皿和PFA杯均用MQ洗净后于7.5 mol/L HNO3中煮沸, 再用MQ水冲洗3~5次, 晾干备用;15 mL或50 mL离心管均用10% HNO3浸泡24 h, MQ水冲洗, 自然晾干备用。
1.2 双氧水氧化元素硒(Se0)的动力学实验取70 mL 0.8 mol/L NaOH+3% H2O2的混合储备液, 加入2.8 mg元素硒(Se0)粉末, 同时放入125 mL血清瓶中, 密封, 快速混均, 放入恒温摇床, 于27℃、150 rpm匀速振荡。分别在0.5 h、1 h、3 h、6 h、12 h、16 h、24 h、36 h时取样, 每次0.25 mL, 并用0.22 μm的无菌滤头过滤。
上述实验中, 由于双氧水具有较强的氧化性, 取样原液获得的不同产物间可能存在继续氧化, 为阻止后续氧化反应的发生, 将所取原液稀释100倍(0.1 mol/L HCl介质)后于4 ℃冰箱保存, 待测。
1.3 硒形态分析和浓度测定 1.3.1 AG1-X8阴离子树脂分离产物元素硒被氧化后, 依据氧化剂氧化性的强弱和介质环境, 可形成不同形态、价态的硒氧阴离子团, 如SeO32-、SeO42-、HSeO3-等。AG1-X8阴离子树脂在中性环境下(Johnson and Bullen, 2004;Clark and Johnson, 2008;Mitchell et al., 2013), 对以阴离子形式存在的Se(Ⅳ)和Se(Ⅵ)离子团具有很强的吸附性, 使用不同浓度的HCl可洗脱出不同价态的Se。为了能准确判定氧化产物形态以及该过程的硒同位素比值变化, 对所取的样品溶液, 均通过AG1-X8阴离子树脂来分离可能形成的Se(Ⅳ)和Se(Ⅵ)。
该分离步骤一般是将体积1 mL的AG1-X8树脂放入洗净的biorad柱中, 用10 mL 6 mol/L的HCl清洗柱子, 然后再用10 mL MQ水淋洗树脂备用;之后取一定体积的样品稀释溶液, 放入PFA杯中, 用0.1 mol/L的HCl或NaOH调节其pH值为中性后样品上柱, 用5 mL MQ水淋洗树脂;再用5 mL 0.1 mol/L的HCl淋洗树脂, 用比色管收集溶液, 此时洗脱出的应为Se(Ⅳ)离子;最后用5 mL 5 mol/L的HCl淋洗树脂, 并用比色管收集含Se(Ⅵ)离子的淋洗液。该流程对Se(Ⅳ)和Se(Ⅵ)的纯溶液而言, Se(Ⅳ)和Se(Ⅵ)的回收率均可达到100%。
1.3.2 氢化物发生器-原子荧光光谱法浓度测试(简称HG-AFS)Se(Ⅳ)、Se(Ⅵ)分离物的测定: 在含Se(Ⅳ)、Se(Ⅵ)阴离子团的淋洗液中, 加入浓HCl调整其介质浓度为5 mol/L HCl, 于90 ℃电热板加热还原55 min, 冷却后备用。取2 mL的还原液, 加MQ水将其介质调整为与HG-AFS的HCl载流液(0.72 mol/L HCl)一致, 待测。HG-AFS工作时的另一股载流质量分数分别为0.2%、1.5%的NaOH和KBH4的混合液。
未分离原液总Se的测定: 取1 mL的稀释原液放入PFA杯中, 加入0.5 mL的浓硝酸, 90℃蒸至20~30 μL左右, 将其用5 mL 5 mol/L的HCl转移到30 mL比色管中, 90 ℃电热板加热还原55 min, 冷却后取2 mL还原液, 其他步骤同上。
2 结果与讨论 2.1 氧化产物的形态判定所有氧化实验的产物浓度数据列于表 1。0.8 mol/L NaOH+3% H2O2混合溶液氧化元素硒(Se0)粉末的实验中, 经AG1-X8树脂分离的原液由HG-AFS进行简单的形态分析。结果表明, 在原液中同时含有Se(Ⅳ)和Se(Ⅵ), 且Se(Ⅳ)、Se(Ⅵ)离子浓度的加和值与未经树脂分离的原液总浓度具有良好的一致性(图 1), 线性相关系数为0.9982, 斜率为1.0244, 说明在误差范围内原始未分离样品的总硒浓度与分离产物Se(Ⅳ)和Se(Ⅵ)离子的浓度加和值相同。另外, 未经树脂分离的原液经浓HNO3加热处理, 这使元素硒在氧化过程可能形成的中间产物(如硒代硒酸根离子)能完全转化为Se(Ⅵ)离子。据此, 也可判定H2O2氧化Se0的过程几乎不会形成除四、六价硒氧离子外的中间产物。
|
|
表 1 双氧水(H2O2)氧化元素硒对应产物浓度数据 Table 1 Concentrations of different products during elemental Se oxidization by hydrogen peroxide(μg/mL) |

|
图 1 未分离产物总Se与分离产物Se浓度之间的关系(H2O2氧化Se0) Figure 1 The relationship between Se contents in unprocessed liquid and the sum of Se(Ⅳ)+Se(Ⅵ)in processed liquid after AG1-X8 seperation |
根据标准条件下氧化还原电势(Eh0)的大小关系(华彤文和杨骏英, 1990;彭安等, 1995): H2O2>SeO42->SeO32->Se0, 即H2O2可氧化元素硒(Se0), 形成四、六价硒氧离子的混合液。由此, H2O2氧化元素硒(Se0)所形成产物的形态分析结果与理论预测结果是一致的。
2.2 双氧水氧化元素硒的动力学模拟27℃下双氧水氧化元素硒的动力学过程如图 2所示, 反应产物的总Se浓度先呈指数快速增大、后期趋于平衡的变化趋势;相较于产物的总Se浓度, Se(Ⅵ)的浓度呈慢速增大、后期稳定的变化规律。Se(Ⅳ)与Se(Ⅵ)浓度变化的规律截然不同, 在起初的3 h内, SeO32-离子迅速生成, 此后不断减少;在反应6 h时, SeO32-和SeO42-的浓度相等, 即两者浓度达平衡。至此, 可推测H2O2氧化元素硒(Se0)可分以下两步反应:

|
图 2 H2O2氧化元素硒的浓度变化曲线 Figure 2 Concentration changing of products in solution during elemental Se oxidization by H2O2 |
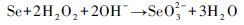
|
(1) |
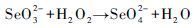
|
(2) |
对该实验反应产物Se浓度的均值(即分离产物浓度加和值与未分离产物浓度的均值)进行动力学模拟(图 3、 表 2), 结果显示, 反应后期, H2O2氧化Se(Ⅳ)的反应符合一级动力学方程;而H2O2氧化元素硒的整体过程可用拟一级动力学方程和拟二级动力学方程描述。所涉及的动力学微积分方程如下(Weston Jr and Schwarz, 1972;Kumar et al., 2006;Lv et al., 2006):

|
图 3 H2O2氧化元素硒的动力学拟合线 Figure 3 Fitted curve of oxidation kinetic equation of elemental Se oxidization by H2O2 |
|
|
表 2 H2O2氧化元素硒的动力学参数表 Table 2 Kinetic parameters of elemental Se oxidization by H2O2 |
一级反应, 微分式为
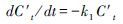
|
(3) |
积分后, 有

|
(4) |
拟一级反应, 微分式为
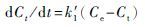
|
(5) |
对其积分, 有
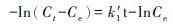
|
(6) |
拟二级反应, 微分式为
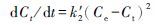
|
(7) |
对其积分, 其表达式变为
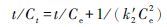
|
(8) |
式中: Ct′为反应物t时刻的浓度(μg/mL), C0为反应物的初始浓度(μg/mL);Ct为生成物在t时刻的浓度(μg/mL), Ce为氧化平衡时生成物浓度(μg/mL);k1为一级氧化速率常数(h-1), k1′为拟一级氧化速率常数(h-1), k2′为拟二级氧化速率常数(mL·μg-1·h-1)。实验拟合结果见图 3和表 2。
产物总Se浓度均值的拟一级动力学拟合方程为(图 3, 表 2): -ln(Ce-Ct)=0.1381t-1.8302, 相关系数R2=0.9349, 拟一级氧化速率常数为0.1381 h-1, 计算的平衡氧化量为6.24 μg/mL, 与实验最大氧化量13.07 μg/mL相差6.83 μg/mL。而由拟二级拟合方程: t/Ct=0.0747t+0.0847, 可以得出t/Ct(h/(μg/mL))与时间t(h)的拟合线相关系数R2高达0.9992, 拟二级氧化速率常数k′ 2为0.0659(mL·μg-1·h-1), 计算的平衡氧化量为13.39 μg/mL, 与36 h时实验获得的氧化量13.07 μg/mL仅相差0.32 μg/mL。因此, H2O2氧化元素硒(Se0)的过程仅生成Se(Ⅳ)和Se(Ⅵ)阴离子团, 且生成Se(Ⅳ)和Se(Ⅵ)混合产物的过程遵循拟二级动力学反应规律, 并在反应36 h后, 该氧化过程近似达到平衡。
另外, 从理论上预测(反应式1和2), 元素硒(Se0)与H2O2完全反应的化学计量比为1 : 3, 实验中这两者的摩尔比约为1 : 2000, H2O2显著过量。一般具有强氧化性的、过量的H2O2可持续氧化元素硒(Se0), 但在氧化实验的后期, 观察到Se(Ⅳ)离子浓度变化不同于Se(Ⅵ)离子且不断减小的奇特现象, 这与理论预测不相符。此外, 反应后期还观察到Se(Ⅳ)离子与Se(Ⅵ)离子的浓度几乎呈镜像变化的现象(图 2), 暗示着H2O2氧化元素硒(Se0)的后期阶段, 以Se(Ⅳ)离子氧化为Se(Ⅵ)离子为主。对3 h后四价硒氧离子的浓度进行动力学模拟(图 3c), 发现-lnct=0.0775t-2.1625, 相关系数R2为0.9929。3 h时, 模拟获得的SeO32-浓度为6.89 μg/mL, 与该时刻SeO32-的浓度7.24 μg/mL只相差0.35 μg/mL, 表明该时刻以后H2O2氧化SeO32-为SeO42-的反应符合一级动力学反应, 速率常数k1为0.0775(h-1), 该特征与化学反应方程式(2)是一致的。由于H2O2氧化元素硒(Se0)的后期, 体系中还存在多种物质(如Se0、SeO32-、SeO42-、H2O2等)和多种界面(如固-液, 液-液), 从单纯的Se(Ⅳ)浓度数据拟合得到的认识很难证实体系本身的特征。为此, 本研究还做了补充实验, 在室温(21±2)℃时, 用0.8 mol/L NaOH+3% H2O2的混合溶液氧化了四价硒(SeO32-)。结果显示(图 4), 反应物Se(Ⅳ)离子浓度的一级动力学拟合线为-lnct=0.0175t-3.8379, 相关系数R2为0.9962, 一级速率常数k1为0.0175(h-1), 模拟获得的反应物初始浓度为46.43 μg/mL, 与初始SeO32-的浓度46 μg/mL相差仅0.43 μg/mL, 进一步表明过剩的H2O2氧化SeO32-为SeO42-的反应符合一级动力学反应, 且是H2O2氧化元素硒后期的主要反应过程。

|
图 4 H2O2氧化亚硒酸钠(Na2SeO3)的浓度变化曲线(a)及动力学拟合线(b) Figure 4 Concentration(a)and fitted(b)curve of oxidation kinetic equation of selenite oxidized by H2O2 |
从元素Se的溶解性, 可知在强碱性条件下, 固体硒(Se0)粉末的表层可溶解活化形成一层犹如电子云状般的硒原子活化层。与内层未溶解活化的硒原子相比, 这种溶解活化的硒原子层所受的束缚力较弱一些, 容易反应, 并对反应速率的大小有一定影响(见反应1及下文论述)。根据上述H2O2氧化元素硒反应动力学模拟的结果, 可知该氧化反应的表观速率(即r=dCt/dt, Ct为t时刻氧化产物的总浓度, μg/mL)可由被氧化的活化硒原子数目(用Ce-Ct表示, Ce为氧化平衡时产物的总浓度, μg/mL)的平方值决定。氧化产物的浓度最终趋于平衡的实验结果表明, 随反应时间增加, (Ce-Ct)值不断减小, 导致整个反应的速率(r)也逐渐变小直至趋于0, 推测这极有可能与元素硒(Se0)微粒的形态、吸附、界面反应等密切有关。
实验所用元素硒(Se0)粉末的南东M图片显示(图 5), 硒粉末多为球形, 以粒径在3~5 μm范围内的球粒为主, 呈团簇状聚集且团聚体中间形成空间通道, 并在单个球粒或团聚体的表面附着有一定量的硒粉小颗粒(粒径大多小于1 μm), 这种现象表明硒颗粒表面具有一定的吸附能力。溶液中H2O2氧化元素硒(Se0)的过程可能经历了H2O2分子先吸附后被还原或吸附和还原同时发生的过程。在大多数情况下, 拟一级动力学方程只能应用于吸附过程的初始阶段而不是整个阶段, 而准二级动力学方程假定限速阶段可能为化学吸附, 适用于很多研究(Kumar et al., 2006)。从元素硒(Se0)被H2O2氧化的过程符合拟二级动力学方程的结果可知, 具有强氧化性的H2O2分子主要通过化学吸附的方式先迅速附着在固体元素硒(Se0)的周围, 在时间上同步或稍微滞后, 吸附剂(Se0)与吸附质(H2O2)分子在吸附界面(表层Se0原子活化层)也迅速发生电子转移, 通过反应式(1)消耗H2O2分子, 快速生成SeO32-离子, 该过程中被吸附的H2O2分子的量控制着反应初期氧化反应的程度。此外, 当H2O2氧化元素硒(Se0)形成硒氧离子后, 部分硒氧离子(SeO32-和SeO42-)可能会被团聚体颗粒固定或吸附在硒微粒的表面, 或进入团聚体形成的空间通道, 并随着反应进行, 使被固定的硒氧离子量不断积累。由于元素硒(Se0)团聚体的表面积及可供容纳外界离子的空间通道是有限的, 随反应的进行, 呈吸附形式的硒氧离子(SeO32-和SeO42-)可通过与早先吸附的H2O2分子相互竞争位点, 或以包裹元素硒(Se0)的形式减小外界H2O2分子与硒(Se0)原子的反应。再者, 根据不同形态Se的Eh-pH特性, 元素硒(Se0)与固定的SeO42-离子可能会在空间通道或接触界面发生化学归中反应, 生成SeO32-离子。从产物的总Se浓度变化规律来看(图 2), 随着被固定的硒氧离子量的增大, 推测元素硒原子的表面最终可能形成了一层硒氧离子隔层, 阻碍过剩的H2O2分子与元素硒(Se0)接触, 使整个反应最终平衡。在氧化反应的后期, SeO42-离子达到平衡暗示着H2O2氧化SeO32-生成SeO42-的反应与元素硒(Se0)还原消耗吸附固定态SeO42-的反应达到了平衡。因此, 可以认为氧化产物SeO32-和SeO42-主要以包裹元素硒(Se0)为主、竞争吸附位点为辅的形式阻断外界H2O2分子与硒(Se0)原子的反应, 进而影响H2O2分子在元素硒(Se0)表面的化学吸附作用, 最终控制整个反应。

|
图 5 元素硒(粉末)的扫描电镜图(南东M) Figure 5 SEM photo of elemental Se(powder) |
由此可知, H2O2氧化元素硒(Se0)的反应是一个相对复杂的反应, 涉及到2个反应并伴有Se(Ⅳ)中间价态离子的生成。反应产物的总Se浓度变化可用拟二级动力学方程来描述的实验事实, 说明H2O2分子、SeO32-以及SeO42-等在元素硒(Se0)团聚体的表面和其所形成的空间通道中, 发生的界面化学吸附作用是整个反应的控制步骤。类比于衰变反应的半衰期, 本实验中可以得出27℃下H2O2氧化元素硒(Se0)的倍增期(Weston Jr and Schwarz, 1972;Wright, 1999)(表 2)为0.57 h, 最终约33%的元素Se0氧化为极易迁移和被生物利用的硒氧离子(以SeO42-离子为主)(表 1)。最后, 27℃时, 从H2O2氧化元素硒(Se0)的动力学拟合参数来看, 反应后期阶段SeO32-被氧化的一级反应速率常数为0.0775 h-1;而室温下(21±2)℃, H2O2氧化亚硒酸钠的一级反应速率常数为0.0175 h-1。前者速率约为后者的4倍, 速率常数的差别极可能由温度的差异引起, 表明该反应受温度的影响较大。
在自然界中, 实际反应的程度与反应物浓度、pH值、温度、反应时间等密切相关, 实验结果在自然体系中的应用还需谨慎。虽然本文的实验条件(强碱性)与自然条件有所不同, 但上述这些参数的获得, 说明元素硒(Se0)与H2O2的反应十分强烈。无论是H2O2氧化元素硒还是亚硒酸钠, 最终均形成迁移性最强且最易被生物利用的Se(Ⅵ)离子, 存在较大的潜在环境风险。由此, 或随雨水进入地面(Penkett et al., 1979, 2007;González-Davila et al., 2005)、或由植物体内部羟基自由基生成, 亦或由高放射性地质体中水辐射分解生成的(Cooper and Lean, 1989;黄贤黎等, 2013)、普遍存在于环境中的H2O2(Pettine et al., 1999), 即使其含量低、反应时间短, 其对还原态元素硒(Se0)的氧化释放过程可能仍具有重要影响, 这也可能是自然界中元素硒罕见发现的原因之一。
3 结论本文通过实验模拟, 研究了元素Se0被H2O2氧化至硒氧离子过程中的反应动力学规律, 并探讨了控制反应过程的可能机制。利用HG-AFS对反应溶液的形态分析表明, H2O2氧化元素硒可形成四价和六价硒氧离子, 反应的结果与通过氧化还原电势理论预测的结果一致。氧化动力学模拟的结果表明, 27 ℃时, H2O2氧化元素硒(Se0)的整个过程可用拟二级动力学方程来描述, 速率常数为0.0659 mL·μg-1·h-1。该反应的动力学机理是比较复杂的, 与元素硒的形态、表面原子活化溶解、化学吸附等密切相关。H2O2分子在元素硒(Se0)表层的化学吸附有可能是H2O2氧化元素硒(Se0)反应的控制步骤。元素硒(Se0)的氧化实验结果表明, H2O2可能存在的氧化作用不容忽视, 尽管实际过程中这种作用的影响程度亟待进一步研究, 但对理解不同环境介质中元素硒的迁移、转化和释放有着重要的指示意义, 特别是对高放射性地质体中硒的无机氧化释放机理具有一定的指导作用。
致谢: 中国科学院地球化学研究所秦海波副研究员为本文的撰写和修改贡献了宝贵意见, 梁良博士、戴余优硕士、赵博硕士对本实验浓度数据的测定提供了无私的帮助, 凌宏文工程师、唐扬工程师在仪器的维护和使用上给予了极大支持, 谨致谢忱。
| [] | Bienvenu P, Cassette P, Andreoletti G, Bé M M, Comte J, Lépy M C. 2007. A new determination of 79Se half-life. Applied Radiation and Isotopes , 65 (3) : 355–364. DOI:10.1016/j.apradiso.2006.09.009 |
| [] | Chen F R, Burns P C, Ewing R C. 1999. 79Se:Geochemical and crystallo-chemical retardation mechanisms. Journal of Nuclear Materials , 275 (1) : 81–94. DOI:10.1016/S0022-3115(99)00105-1 |
| [] | Clark S K, Johnson T M. 2008. Effective isotopic fractionation factors for solute removal by reactive sediments:A laboratory microcosm and slurry study. Environmental Science & Technology , 42 (21) : 7850–7855. |
| [] | Cooper W J, Lean D R S. 1989. Hydrogen peroxide concentration in a northern lake:Photochemical formation and diel variability. Environmental Science & Technology , 23 (11) : 1425–1428. |
| [] | Dumont E, Vanhaecke F, Cornelis R. 2006. Selenium speciation from food source to metabolites:A critical review. Analytical and Bioanalytical Chemistry , 385 (7) : 1304–1323. DOI:10.1007/s00216-006-0529-8 |
| [] | Finkelman R B, Belkin H E, Zheng B S. 1999. Health impacts of domestic coal use in China. Proceedings of the National Academy of Sciences of the United States of America , 96 (7) : 3427–3431. DOI:10.1073/pnas.96.7.3427 |
| [] | Fordyce F. 2007. Selenium geochemistry and health. AMBIO:A Journal of the Human Environment , 36 (1) : 94–97. DOI:10.1579/0044-7447(2007)36[94:SGAH]2.0.CO;2 |
| [] | González-Davila M, Santana-Casiano J M, Millero F J. 2005. Oxidation of iron(Ⅱ)nanomolar with H2O2 in seawater. Geochimica et Cosmochimica Acta , 69 (1) : 83–93. DOI:10.1016/j.gca.2004.05.043 |
| [] | Grambow B. 2008. Mobile fission and activation products in nuclear waste disposal. Journal of Contaminant Hydrology , 102 (3-4) : 180–186. DOI:10.1016/j.jconhyd.2008.10.006 |
| [] | Herring J R. 1991. Selenium geochemistry-a conspectus. In:Severson R C, Fisher Jr S E, Gough L P eds. Proceedings of the 1990 billings land reclamation symposium on se in arid and semiarid environments, western United States. Denver:US Geological Survey Circular , 1064 : 7–23. |
| [] | Johnson T M, Bullen T D. 2004. Mass-dependent fractionation of selenium and chromium isotopes in low-temperature environments. Reviews in Mineralogy and Geochemistry , 55 (1) : 289–317. DOI:10.2138/gsrmg.55.1.289 |
| [] | Kang M L, Ma B, Bardelli F, Chen F R, Liu C L, Zheng Z, Wu S J, Charlet L. 2013. Interaction of aqueous Se(Ⅳ)/Se(Ⅵ)with FeSe/FeSe2:Implication to Se redox process. Journal of Hazardous Materials , 248-249 : 20–28. DOI:10.1016/j.jhazmat.2012.12.037 |
| [] | Kumar Y P, King P, Prasad V S R K. 2006. Equilibrium and kinetic studies for the biosorption system of copper(Ⅱ)ion from aqueous solution using Tectona grandis L. f. leaves powder. Journal of Hazardous Materials , 137 (2) : 1211–1217. DOI:10.1016/j.jhazmat.2006.04.006 |
| [] | LÜ L, He J, Wei M, Evans D G, Duan X. 2006. Uptake of chloride ion from aqueous solution by calcined layered double hydroxides:Equilibrium and kinetic studies. Water Research , 40 (4) : 735–743. DOI:10.1016/j.watres.2005.11.043 |
| [] | Ma B, Nie Z, Liu C L, Kang M L, Bardelli F, Chen F R, Charlet L. 2014. Kinetics of FeSe2 oxidation by ferric iron and its reactivity compared with FeS2. Science China Chemistry , 57 (9) : 1300–1309. DOI:10.1007/s11426-014-5126-7 |
| [] | Mitchell K, Couture R M, Johnson T M, Mason P R D, Van Cappellen P. 2013. Selenium sorption and isotope fractionation:Iron(Ⅲ)oxides versus iron(Ⅱ)sulfides. Chemical Geology , 342 : 21–28. DOI:10.1016/j.chemgeo.2013.01.017 |
| [] | Penkett S A, Jones B M R, Brice K A, Eggleton A E J. 2007. The importance of atmospheric ozone and hydrogen peroxide in oxidising sulphur dioxide in cloud and rainwater. Atmospheric Environment , 41 (S) : 154–168. |
| [] | Penkett S A, Jones B M R, Brich K A, Eggleton A E J. 1979. The importance of atmospheric ozone and hydrogen peroxide in oxidising sulphur dioxide in cloud and rainwater. Atmospheric Environment(1967) , 13 (1) : 123–137. DOI:10.1016/0004-6981(79)90251-8 |
| [] | Pettine M, Campanella L, Millero F J. 1999. Arsenite oxidation by H2O2 in aqueous solutions. Geochimica et Cosmochimica Acta , 63 (18) : 2727–2735. DOI:10.1016/S0016-7037(99)00212-4 |
| [] | Schilling K, Johnson T M, Mason P R D. 2014. A sequential extraction technique for mass-balanced stable selenium isotope analysis of soil samples. Chemical Geology , 381 : 125–130. DOI:10.1016/j.chemgeo.2014.04.014 |
| [] | Tan J A, Zhu W Y, Wang W Y, Li R B, Hou S F, Wang D C, Yang L S. 2002. Selenium in soil and endemic diseases in China. Science of the Total Environment , 284 (1-3) : 227–235. DOI:10.1016/S0048-9697(01)00889-0 |
| [] | Thompson M E, Roach C, Braddock W. 1956. New occurrences of native selenium. American Mineralogist , 41 (1-2) : 156–157. |
| [] | Weston Jr R E, Schwarz H A. 1972. Chemical kinetics. New Jersey: Prentice-Hall . |
| [] | Winkel L H E, Johnson C A, Lenz M, Grundl T, Leupin O X, Amini M, Charlet L. 2012. Environmental selenium research:From microscopic processes to global understanding. Environmental Science & Technology , 46 (2) : 571–579. |
| [] | Wright W G. 1999. Oxidation and mobilization of selenium by nitrate in irrigation drainage. Journal of Environmental Quality , 28 (4) : 1182–1187. |
| [] | Zhu J M, Johnson T M, Clark S K, Zhu X K, Wang X L. 2014. Selenium redox cycling during weathering of Se-rich shales:A selenium isotope study. Geochimica et Cosmochimica Acta , 126 : 228–249. DOI:10.1016/j.gca.2013.11.004 |
| [] | Zhu J M, Johnson T M, Finkelman R B, Zheng B S, Sýkorová I, Pešek J. 2012. The occurrence and origin of selenium minerals in Se-rich stone coals, spoils and their adjacent soils in Yutangba, China. Chemical Geology , 330-331 : 27–38. DOI:10.1016/j.chemgeo.2012.08.023 |
| [] | Zhu J M, Wang N, Li S H, Li L, Su H C, Liu C X. 2008. Distribution and transport of selenium in Yutangba, China:Impact of human activities. Science of the Total Environment , 392 (2-3) : 252–261. DOI:10.1016/j.scitotenv.2007.12.019 |
| [] | Zhu J M, Zuo W, Liang X B, Li S H, Zheng B S. 2004. Occurrence of native selenium in Yutangba and its environmental implications. Applied Geochemistry , 19 (3) : 461–467. DOI:10.1016/j.apgeochem.2003.09.001 |
| [] | 华彤文, 杨骏英. 1990. 普通化学原理. 化学教育 , 11 (1) : 27. |
| [] | 黄贤黎, 杨波, 刘义保. 2013. 水的辐射分解过程和产物及其对环境的影响. 能源研究与管理 (4) : 32–35. |
| [] | 彭安, 王子健, WhangerP D. 1995. 硒的环境生物无机化学. 北京: 中国环境科学出版社 . |
| [] | 秦海波, 朱建明, 李社红, 雷磊, 尚林波. 2008. 环境中硒形态分析方法的研究进展. 矿物岩石地球化学通报 , 27 (2) : 180–187. |
| [] | 张莹, 刘桂建, 郑刘根, ChouC L, 齐翠翠. 2007. 中国煤中硒的环境地球化学. 矿物岩石地球化学通报 , 26 (4) : 389–398. |
| [] | 朱建明, 雷磊, 肖湘, 袁永强, 秦海波, 苏惠. 2011. 地衣芽孢杆菌对亚硒酸盐的还原. 矿物岩石地球化学通报 , 30 (3) : 245–250. |