 2016, Vol. 35
2016, Vol. 35



Cu有2个同位素:63 Cu和65 Cu,它们在自然界的丰度分别为69.17%和30.83%(Shields et al.,1965),常以不同的价态(0,+1,+2)赋存于各类岩石、矿物、流体和生物体中,并广泛参与各种地球化学和生物化学过程(王跃和朱祥坤,2010)。Cu同位素作为一种新的地球化学示踪手段,为揭示自然界中各类地质作用、矿床成因及分布规律、生物地球化学循环过程等提供了新的证据。上世纪五六十年代,Walker等(1958)利用表面热电离质谱仪对天然样品中的Cu同位素进行了测定,但由于测定过程中同位素的歧视效应太大,未能得到有意义的同位素比值,因而无法通过同位素的组成探讨样品的地质意义。近年来,同位素分析方法的改进和新一代多接收等离子质谱仪(MC-ICP-MS)的应用使得Cu同位素的分析误差达到小于0.04‰的水平,实现了Cu同位素的高精度测试(Albarède,2004;Takano et al.,2013)。
Maréchal等(1999)首次对不同矿物和生物样品的δ65 Cu 进行了测定,率先在铜同位素组成的研究上取得了新的突破,为Cu同位素测定的化学前处理方法奠定了基础,并利用多接收双聚焦等离子质谱(VG Plasma 54)克服了传统ICP源不稳定的影响,对Cu同位素组成进行了精确测定。此后,Archer和Vance(2004)通过改进Maréchal等(1999)的化学分离和质谱测试方法,实现了对较少样品量的Cu、Zn同位素的高精度测试,并讨论了Fe、Sr等基质元素以及Cu/Zn的变化对目标元素的影响。Borrok等(2007)对金属元素浓度变化大、基质元素含量高的复杂水系中的Cu、Fe、Zn同位素的分离和质谱测试方法进行了研究,为识别淡水系统中金属元素的来源以及生物地球化学过程提供了重要研究手段。除此之外,对海水中过渡族元素同位素的测定也受到了很多学者的关注,但盐的含量高和目标元素浓度低两大因素制约了海水中同位素应用,Wu和Boyle(1997)以及Saito和Schneider(2006)用Mg(OH)2共沉淀法对海水进行了预富集的研究,使海水中低含量的金属元素的测定成为可能。Bermin等(2006)和Vance等(2008)较早对海水中的Cu同位素进行了测定,为后续学者的研究提供了较完善的测定方案和较系统的海水δ65 Cu的数据。
近些年,中国学者也开展了Cu同位素的研究工作。蒋少涌等(2001)对热液硫化物矿床的Cu同位素进行了研究;蔡俊军等(2006)对不同矿床的Cu同位素进行分析,不仅在Cu同位素样品的选择、制备和分析方法方面积累了丰富的经验,并且对分馏机制也进行了探讨;梁莉莉等(2006)对Cu、Zn同位素方法在环境地球化学研究中的应用进行了探讨和展望;唐索寒等(2006)对地质样品中Cu、Fe、Zn同位素的标准物质进行了纯化研究;侯可军等(2008)建立了高精度的Cu、Zn同位素测试技术;Liu等(2014)建立了高精度Cu、Fe同位素的测试方法,外部精度可达±0.05‰,分析精度的提高证实了火成岩中存在Cu同位素分馏(火成岩的δ65 Cu变化范围为-0.01‰~0.39‰),为示踪高温环境下的地质过程提供了有效的证据。
目前,Cu同位素的测试主要存在2方面的问题:①仪器的质量歧视,需要进行一定的校正,目前主要有SSB法、内标法、双稀释剂法及一些在以上方法的基础上进行改进或组合形成的校正方法;②需要一个标准样品作为基准值,而Cu同位素的国际认可标样NIST SRM 976已经停产,因此急需一种新的Cu同位素标样。
Hou等(2016)在前人研究的基础上,对Cu同位素的化学分离流程进行了改进,解决了目前Cu同位素测试中遇到的2大问题:①以Ga作内标,用内标法与SSB法的结合法(C-SSBIN)校正仪器的质量歧视,精度较传统的直接SSB法提高了5倍左右;②推荐了一种新的Cu同位素标样NIST SRM 3114,准确给出了NIST SRM 3114中铜同位素的组成,即65 Cu/63 Cu=0.4470±0.0013,并得到了其相对原有标样NIST SRM 976的转换值0.18±0.04‰,为铜同位素比值测定提供了新的标准。本文在总结前人成果的基础上,结合笔者新近的工作,对Cu同位素的化学前处理以及测试方法进行了综述,以期为Cu同位素在相关领域的研究提供资料。
1 铜同位素的表示方法及标准物质 目前,Cu同位素的表示方法一般采用δ值法,即样品的同位素比值相对于某一标准样品的同位素比值的千分偏差。NIST SRM 976是现在国际认可的Cu同位素标样,不同实验室所报道的数据都是相对于NIST SRM 976的比值,但目前该标样已经停产,各实验室需要根据自身情况选择合适的替代标样,并得到其相对国际认可标样的转换值,使测试数据有统一的基准点。Moeller等(2012)对IRMM 生产的ERM-AE633和ERM-AE647进行了研究,给出了这两种标样相对于NIST 976的δ65 Cu值分别为0.01‰±0.05‰、0.21‰±0.05‰;Liu等(2014)选择GSB Cu作为实验室内部标样,并得到其相对NIST 976的δ65 Cu值为0.44‰±0.04‰。这两个报道虽然给出了替代标样相对原有标样的δ65 Cu值,但均未涉及所推荐标样的绝对同位素比值。Hou等(2016)通过研究NIST公司的SRM 3114样品,不仅得到了其相对原有标样NIST 976 Cu的δ65 Cu值0.18‰±0.04‰,还给出了NIST 3114 Cu的绝对同位素比值65 Cu/63 Cu=0.4470±0.0013,因此,NIST 3114作为Cu的候选标准样品之一,更具有竞争性和优越性。
2 样品的纯化及分离自然界中Cu的含量差别很大,大陆地壳中Cu含量的平均值约为27 μg/g(Rudnick and Gao,2003),大洋地壳为86 μg/g(Taylor and McLennan,1985),亏损地幔为30 μg/g(McDonough,2003),地核为125 μg/g(McDonough,2003)。不同岩石样品的Cu含量也有一定的差异,各类岩石标样中Cu含量的变化范围为20~200 μg/g(李丹丹,2015)。海水中Cu的含量一般低于5 nmol/L(Vance et al.,2008;Takano et al.,2013;Thompson and Ellwood,2014),河水则可达到5~60 nmol/L(Vance et al.,2008)。为了确保Cu同位素的分析测试精度,一般都需要对样品进行预处理,对含量较低的样品(如海水)还需要进行预富集(Wu and Boyle,1997; Bermin et al.,2006;Saito and Schneider,2006;Vance et al.,2008)。样品的预处理一般包括消解和纯化,常见的消解方法包括湿法消解(李世珍等,2011;王家松和彭丽娜,2012)和干法灰化(李世珍等,2011),纯化分离则关系到样品测试的准确性,很多学者都在Maréchal等(1999)的基础上对此进行了研究(Korkisch and Worsfold,1990;唐索寒和朱祥坤2006; 唐索寒等,2006;Borrok et al.,2007;马信江等,2009;Liu et al.,2014;Hou et al.,2016)。纯化后的样品可以根据需要进行稀释,通常进样浓度在0.2~0.5 μg/g范围内时可获得最佳测试效果(Hou et al.,2016)。
Cu同位素比值的测定可能会受到同质异位素的干扰,如23Na 40 Ar+、23Na162OH+、64ZnH+、28Si 35 Cl+、30Si 35 Cl+、28Si 37 Cl+、 130 Ba2+、33S16O+2等,只有将Cu元素有效地从基质元素中分离出来,才能避免这些离子的干扰,从而准确测定Cu同位素比值;其次,还要注意分离过程中Cu同位素的回收率,只有100%的回收才能有效避免在分离纯化过程中因分馏而导致的结果失真。分离纯化Cu同位素时一般采用离子交换层析法,早期分离时较常使用的是AG1强碱型阴离子交换树脂,但该树脂不能有效地保留Cu,从而无法使Cu与基质元素完全分离(Maréchal et al.,1999),van der Walt等(1985)发现大孔径强碱型氯型阴离子交换树脂AGMP-1(200~400目)在浓HCl条件下对Cu2+、Fe3+和Zn2+的分配系数比AG1离子交换树脂高,可以有效地分离铜与其他基质元素,为后续学者的研究奠定了基础。Maréchal等(1999)最先使用AGMP-1树脂对Cu等元素进行分离,该树脂的活性基团为R-CH2N+(CH3)3,在HCl溶液中易与Cl-结合,而Cu2+在浓HCl溶液中易与Cl-结合形成络合离子CuCl+、CuCl2、CuCl-3和CuCl2-4,这些络合离子在交换柱上与树脂中的Cl-发生交换,根据不同的络合离子与树脂亲和力的差异,选择不同种类的酸和合适的酸度,可以将Cu等元素分离出来。Maréchal等(1999)以Ramette(1986)的淋洗模型为依据,对Cu、Fe、Zn的分离过程作了详细的研究,并使用7 mol/L HCl、2 mol/L HCl和0.5 mol/L HNO3对目标元素进行过柱分离,为后来者的研究奠定了重要的基础(Mason et al.,2005;唐索寒和朱祥坤,2006; 唐索寒等,2006; 孙剑等,2009;Pribil et al.,2010)。
唐索寒等(2006)认为,在Maréchal等(1999)的过柱条件下,Cu和Co的洗脱峰几乎完全重叠,不能实现Cu和Co的有效分离,并通过一系列实验测定发现,在m(Co/Cu)<10时,Co对Cu同位素比值的测定无影响,而一般地质样品中m(Co/Cu)<7(赵振华,1992)(如,闪锌矿约为0.5,闪长岩约为2),所以该方法可以满足大多数地质样品中Cu同位素的分离要求。对于部分Co含量高的样品,唐索寒和朱祥坤(2006)根据树脂亲和力与溶液酸度相关的原理,通过改变酸的浓度(在6 mol/L HCl)可以有效地分离Cu与Co,因此,建议对高Co含量的部分硅酸盐等样品,可以先用7 mol/L HCl将Cu、Co一起洗脱,然后再用6 mol/L HCl实现Cu、Co的分离;而对于一些基质元素含量较少的样品,如硫化物、氧化物、氢氧化物等,可以直接用 6 mol/L HCl将Cu与包括Co在内的基质元素进行有效地分离。Maréchal(2002)和Albarède(2002)研究发现,Cu同位素在经过AGMP-1树脂时会发生微弱的同位素分馏,样品分离次数越多产生的分馏越大,对测定同位素的比值影响也会越大。因此,很多研究者在此基础上对Cu同位素的分离方法进行了改进,使Cu与Co只需要经过一次交换柱就可以实现完全分离(Borrok et al.,2007;Liu et al.,2014;Hou et al.,2016)。
Borrok等(2007)认为,Maréchal等(1999)的分离方法虽然经典,但在淋洗过程中消耗了大量的酸(51 mL),会导致本底升高,且部分Cu与基质元素无法在第一次交换柱后完全分离,必须进行第二次分离才能达到分析要求。因此,Borrok等(2007)在该经典流程的基础上,将淋洗杂质的过程中所用HCl浓度由7 mol/L提高为10 mol/L,这样只需进行一次交换柱分离,就可以很好地除去基质元素,之后又改用5 mol/L HCl洗脱Cu元素,整个流程淋洗分离Cu的过程所用酸不超过15 mL,在降低本底的同时也缩短了交换柱分离的时间。
Korkisch和Worsfold(1990)曾报道,在8~9 mol/L HCl介质下,Co2+的络合物与强碱性阴离子树脂间的分配系数大于Cu2+的络合物。因此,理论上讲,在该酸度条件下可以一次性将Cu与包括Co在内的基质元素进行有效地分离。
马信江等(2009)较早用新型阴离子树脂AGMP-1M(100~200目,活性基团与AGMP-1树脂相同)进行了尝试,发现在6~8.2 mol/L HCl作介质的条件下,随着HCl浓度的增加,Cu与Co的淋洗曲线重叠部分逐渐变少,但Cu的拖尾现象较严重,导致Co的淋洗液中出现了少量的Cu,降低了回收率。这可能与HCl浓度增加时生成了少量的CuCl3-5有关,因为CuCl3-5的亲和力大于同时存在的CuCl-3和CuCl2-4的亲和力,所以HCl浓度的增加会导致Cu的拖尾现象(Brugger et al.,2001)。马信江等(2009)在HCl中加入0.01%(体积)HF,F-可以取代CuCl-3、CuCl2-4和CuCl3-5中的Cl-,同时抑制CuCl3-5的产生,使Cu的淋洗曲线提前出现且明显变陡,从而实现了Cu与Co的彻底分离,提高了Cu元素的回收率,并且仅通过一次交换柱就可以将Cu与包括Co在内的基质元素分离,提高了分离效率。
Liu等(2014)也选用AGMP-1M树脂对Cu、Fe的分离进行了研究,实验中将Maréchal等(1999)淋洗Cu的过程中使用的7 mol/L HCl改为8 mol/L HCl,可以将Na、Mg、Ti、Co等基质元素与Cu进行有效地分离,在酸中加入H2O2可以使样品中的Cu、Fe氧化为最高价态,从而避免纯化过程导致的分馏,最终得到Cu的回收率在100%左右。
Hou等(2016)提出的分离Cu的方法简单高效。实验中用8.5 mol/L HCl代替7 mol/L HCl,获得的淋洗液中Co/Cu小于0.02,实现了Cu和Co的有效分离;在淋洗液中加入少量HF,实现了Cu和Ti的分离并减少了Cu的拖尾。该方法的优点是:①耗酸少,降低了分离过程的本底;②只需一次交换柱就可以将Cu从基质元素中较好地分离出来,且回收率也能到达100%左右。
3 基质元素的干扰评估经过纯化分离后的样品,大多数基质元素都被洗脱出去,但一些含量较高的元素(如Na、Fe等)和较难分离的元素(如Co等)则可能残留较多。这些残留的元素可能会在Cu同位素的测定过程中产生多原子干扰(如23 Na 40 Ar+,23 Na162O1H+,47Ti16O+,46 Ti16O1H+等对63 Cu,49Ti16O+,23Na182O1H+,48Ti16O1H+对65 Cu),还有可能引起基质效应,如果残留过多则会影响测定结果的准确性,所以有必要对纯化后样品中的基质元素所引起的干扰进行评估。
Na是一种常见的主量元素,由Na产生的多原子(如23Na 40 Ar+)会对63 Cu产生较大的干扰。Liu等(2014)认为在Na/Cu值小于1时,标样的分馏值在误差范围内,即此时Na对δ65 Cu值的测定没有影响。 Hou等(2016)发现,当Na/Cu值小于0.5时,对δ65 Cu测定值的影响可以忽略,而作者用8.5 mol/L HCl分离后的样品溶液中Na/Cu值小于0.05,因此Na离子形成的多原子干扰问题可以忽略。
地壳中Fe的丰度高达5.8%,是Cu丰度(0.055‰)的1000倍以上,经化学纯化后可能仍有少量的Fe未被完全分离。现有的各种报道对Fe的干扰评估意见并不一致。蔡俊军等(2006)研究发现,当Fe/Cu值在0.1~100时,Fe对Cu同位素的测定没有影响,这与Zhu等(2000)的报道一致。而侯可军等(2008)的研究却发现,当Fe/Cu<5时测定的δ65 Cu值才与报道值一致。Liu等(2014)的报道则显示,当Fe/Cu<1时,对δ65 Cu值的测定无影响。Hou等(2016)研究发现在Fe/Cu值<25时,可以忽略Fe的影响,而作者测定其纯化后的Cu样品溶液中Fe/Cu<15,因此在测试时可以忽略Fe的影响。
Co是一种较难与Cu分离的元素,Maréchal等(1999)的分离方法中,Cu的接收液中都含有少量的Co。唐索寒等(2006)认为当Co/Cu<10时,Co对Cu同位素比值的测定无影响;侯可军等(2008)研究发现当Co/Cu<20时,测定的δ65 Cu值与标准值在误差允许的范围内一致;Hou等(2016)发现当Co/Cu<1时,可以忽略Co对Cu同位素测定造成的影响,其改进的分离方法可以满足此要求。
Ti有5个同位素,对Cu同位素的测定存在多原子干扰(如47Ti16O+、46Ti16O1H+对63 Cu,49Ti16O+、48Ti16O1H+对65 Cu),基于Ti各同位素的丰度比,Hou等(2016)初步估计了这些干扰可能造成的影响:首先,因为47Ti(7.44%)的丰度高于49Ti(5.41%),所以Ti的氧化物(47Ti16O+)造成的干扰可能会降低65 Cu/63 Cu值;而 48Ti(73 . 72%)的丰度远高于46 Ti(8.25%),所以Ti的氢氧化物(48 Ti16 O1H+)造成的干扰可能会使65 Cu/63 Cu值升高。总的来说,造成偏重干扰的48Ti(73 . 72%)比造成偏轻干扰的47 Ti(7.44%)丰度高很多,所以Ti的残留可能会导致δ65 Cu值的升高,这与作者实验测得的结果一致,且基质Ti对Cu同位素测定的影响非常明显,仅在Ti/Cu<0.1时没有影响。Hou等(2016)经纯化得到的部分样品的Ti/Cu可达0.6,此时的溶液测定可能造成结果的不准确性,笔者采用了基体匹配法(Baker et al.,2004;Craddock et al.,2008;Ikehata et al.,2008)消除这种影响,具体操作是在标准溶液中掺入基质Ti,使其Ti的浓度与样品中所含Ti的浓度匹配,测定结果表明:当Ti/Cu<2时,Ti的影响可以忽略,所以部分Ti/Cu值在 0.1~2时的样品可以用该方法进行测定。
4 仪器质量歧视校正MC-ICP-MS的应用虽然极大地提高了Cu同位素的分析精度,但在测试过程中产生的质量歧视却不容忽略,在过去的几十年里,多种质量歧视校正方法已经被成功应用,主要有双稀释剂法、标准-样品-标准交叉法(SSB法)和内标法(Albarède et al.,2004)。其中双稀释剂法既可以监视化学分离过程的分馏,又可以校正仪器漂移引起的分馏,但需要4个或者4个以上的同位素,适用于Pb(Thirlwall,2000;Kuritani and Nakamura,2003;Fletcher,2007)、Mo(Zhou et al.,2007,2011,2012)等有多个同位素的元素的校正,而Cu只有2个同位素,并不适合用双稀释剂法来进行校正。目前,Cu同位素的校正主要用SSB法和内标法,下面将简单介绍一下这2种校正法及它们结合使用的方法。
4.1 标样-样品-标样交叉法(SSB法)此方法的测定顺序是标样、样品交替进行(即标样-样品-标样-样品……),原理是用仪器对样品前后两个标准溶液的质量歧视来校正仪器对样品的质量歧视,使用该方法的前提是在测试过程中仪器对样品和标样的质量歧视效应相同。其中,标准溶液的“真实”同位素比值Rstd为已知,rstd为测定值,根据这2个值计算出标样的分馏系数k,由前后两个标样的分馏系数的平均值 作为中间所夹样品的分馏系数,再用这个分馏系数k和样品的测定值rsample计算出样品的“真实”同位素比值Rsample,如公式(1):

本文用δ作为同位素比值的表示方法,即:

其中,Rsample是样品的同位素比值, 是样品前后两个标样真实比值的平均值。
是样品前后两个标样真实比值的平均值。
在用SSB法校正的情况下,将公式(1)代入(2)中计算得到:
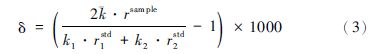
一般情况下,仪器在短时间内是稳定的,时间漂移可以忽略,那么在一个流程内的前后两个标样和中间样品的分馏系数可认为近似相等,即k1=k2= ,此时公式(3)可以简化为:
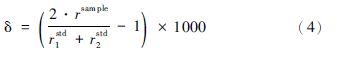
计算过程发现,使用SSB法校正的关键在于样品与标样之间的完美匹配,包括酸度、浓度、基质等方面,以保证标样与样品间分馏系数的可替代性。Zhu等(2000)研究了用SSB法测试Cu同位素的重复性,用Romil Cu作为样品,NIST SRM 976作为标样,2周内的分析精度为0.6ε(2SD)。由此可知,当标样与样品间基体匹配时,SSB法可以校正仪器的质量歧视,但是这种方法并不能对仪器短时间内波动引起的质量歧视进行校正。
4.2 内标法内标法是在样品溶液中加入与被测元素质量数相近的元素,所加入的内标元素的比值是不变的或有国际认可的推荐值,然后用内标元素的分馏系数校正待测元素,从而得出待测元素的比值。该校正方法应用的前提是仪器对样品和标样有相同的质量歧视,目前已经有多种质量歧视校正模型,如线性规律,指数律、幂定律(Ingle et al.,2003;Pichat et al.,2003)、指数定律、一阶和二阶质量歧视校正等,其中,由Russell等(1978)为TIMS测量提出的幂次定律,在MC-ICPMS分析中得到了广泛的应用。常用的内标法可根据选择内标元素的不同细分为以下几种方式(Yang,2009):
(1)内标为被测元素自身的一对同位素。前提是作为内标的同位素对的比值在自然界中是不变的,目前这种方法在Sr(Barbaste et al.,2002)、Nd(Foster and Vance,2006)、Os(Nowell et al.,2008)等元素的同位素分析中广泛应用,如Sr的同位素测试中,利用自然界中比值不变的 88 Sr/86 Sr 校正 87 Sr/86 Sr。
(2)内标元素为其他元素的一对同位素。是内标元素的同位素对的比值具有国际认可的推荐值,此外,在选取内标元素时还应遵循以下原则,即待测样品中不含所加入的内标元素,且内标元素与待测元素质量数相近,电离能相近,化学性质相近等。一般可以选择在元素周期表中与待测元素相近或相邻的元素,如Pb和Tl(Gallon et al.,2008),Tl和Hg(Zheng et al.,2007)等互为内标即是选取了元素周期表中相邻的元素,而Zr和Mo(Anbar et al.,2001),Zr和Rb(Waight et al.,2002)等则是选取了相近的元素作内标。对Cu同位素来说,与其相邻的Zn(Gélabert et al.,2006;侯可军等,2008)和Ni(Graham et al.,2004)是目前普遍采用的内标元素,Ehrlich等(2004)认为,相对于Zn来说,Ni更适合做Cu同位素测定时的内标,Ni(7.64 eV)与Cu(7.73 eV)的第一电离电位比Zn(9.39 eV)与Cu(7.73 eV)的更接近,且分析精度也由0.48‰ 提高到0.16‰。但是,选择第一电离电位相近的元素作为内标元素并不是绝对的,如Pb同位素的测试过程可选Tl作内标,且已得到广泛应用,但是二者的第一电离能却并不相近。Hou等(2016)第一次使用 Ga作为Cu同位素测量的内标,与Zn作内标相比,该方法减少了多原子离子的干扰,并且没有同质异位素的干扰。
(3)线性回归内标法。线性回归内标法是内标法的一种应用形式,同样是在样品溶液中加入已知同位素对比值推荐值的内标元素,目前已经应用于Fe(Kehm et al.,2003),Hg(Xie et al.,2005;Yang and Sturgeon,2009),Ge(Yang and Meija,2010),Ag(Yang et al.,2010)等同位素的测定。与传统的SSB法和内标法相比,线性回归法的最大优点是它可以获得目标同位素比值的绝对值,从而成功得到一种新的具有绝对同位素比值的标样,而不局限于以往的只能获得相对同位素比值的标样。
Hou等(2016)首次以Ga作内标,选用线性回归法(即线性回归内标法)来测定Cu同位素,在元素周期表上,Ga的原子序数为31,与原子序数为29的Cu相近。前人的研究中,大多选用与Cu相邻的Zn或者Ni作内标,与它们相比,选择Ga作内标有以下优点:①Ga的同位素组成简单,只存在2个稳定同位素:69Ga和71Ga,这2个同位素引起的同质异位素和基质效应的干扰相对较小;②Ga在地质样品中的含量相对较少,容易分离,符合样品中不含内标元素的原则,尤其对于含Zn量高的样品,如闪锌矿等,内标元素Ga的应用更具有其独特的优势;③目前使用的截取锥和样品锥多为Ni锥,存在较高的基质效应,而内标Ga可以避免这种基质效应;④内标元素选用的前提条件是具有国际认可的推荐值,而国际标样NIST SRM 994 Ga有高精度的国际认可的推荐值,其同位素比值69Ga/71Ga为1.50676(Machlan et al.,1986)。
线性回归方程可以通过一系列的Rusell方程转化得到,中间不存在任何假设,此处以Cu作为目标元素,Ga作内标元素进行说明:
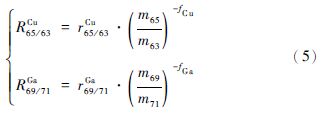
其中: r和R分别为测量值和校正过的“真值”,f是分馏系数,m是核素的相对原子质量。分别对Cu和Ga的分馏方程两边取对数可得:
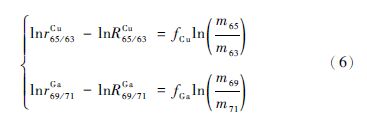
将方程(6)整理后可得到一个有关Cu、Ga同位素比值的对数的线性方程:
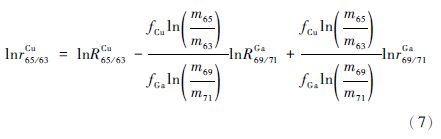
以lnr69/71Ga对lnr65/63Cr作图(图 1),用b代表获得的斜率,a代表截距,那么公式(7)中的斜率b和截距a可以表示为:
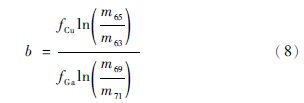
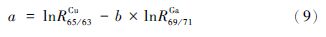

|
图 1 6 h内测定的Cu、Ga同位素比值的对数值的相关性(Hou et al.,2016) Fig. 1 Temporal drift of the copper and galliumisotope ratios during a 6 hr measurement session(Hou et al.,2016) |
每得到一组Cu、Ga同位素的测量值,代入公式(7)便可得到一组斜率与截距,将得到的截距a和斜率b以及推荐的Ga同位素的比值代入方程(9),可以获得Cu同位素的绝对比值R65/63Cu。
由式(7)可以看出,这种校正方法的前提是fCu/fGa在一段测定时间内为常数,通过对测得的待测元素同位素比值与内标元素同位素比值的对数值作图,即可得到被测元素的同位素比值的真值。值得注意的是,这种方法虽然能保证准确度,但是精确度相对较差,为了得到高准确度和高精确度的同位素比值,需要对大量数据进行分析,从中选取二者相关性系数较高的数据作图,再通过计算得到目标元素的同位素比值。Hou等(2016)通过此方法首次准确给出了NIST SRM 3114 Cu标样中Cu同位素比值的绝对值,即65 Cu/63 Cu=0.4470±0.0013(2SD,n=37),并推荐将NIST SRM 3114 Cu作为一种新的Cu同位素标准样品来替代已经停产的NIST SRM 976,为Cu同位素的测定提供了新的基准点。
4.3 内标法与SSB法的结合法内标法与SSB法的结合法,即在样品和标样中加入同等浓度的内标,按“标样-样品-标样”的顺序进行测定,Hou等(2016)首次以Ga作内标,使用该方法对Cu同位素测定过程中的质量歧视进行校正。此方法结合了2种方法的优点,可以对仪器短时间内的漂移进行有效的监测和校正,以下模型计算过程中均以Cu作为待测元素,Ga作为内标元素进行推导,其中被测元素Cu的标样具有国际认可的推荐值,根据Cu同位素标样的推荐值和测定值,以Rusell方程为原型,可以计算出标样中Cu的分馏系数fCustd:

式中: R为推荐值,r为测定值,m为元素的相对原子质量。
Hou等(2016)在计算过程中使用的元素相对原子质量以IUPAC 2003版(de Laeter et al.,2003)为参考,其中63 Cu和65 Cu的精确质量分别为62.9295975和64.9277895,69Ga和71Ga的精确质量分别为68.9255736和70.926022。尽管已经有越来越多的证据表明,在测试过程中,被测元素与内标元素的分馏形式不完全相同,即二者的分馏系数不同,但这种变化是很小的(Maréchal et al.,1999),尤其是在短时间内,或一个测试小组内(标样-样品-标样),因此可以认为fCr≈fGa,所以可以通过Cu的分馏系数fCustd与内标Ga的测定值计算得到标样中Ga的“真值”Rstd69/71:

通过计算前后2个标样中Cu的分馏系数,可以得到前后两个标样中Ga的“真值”,取其平均值得到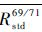 ,这个平均值可以当作2个标样所夹样品中Ga元素比值的真值,从而可以计算出样品中Ga的分馏系数fsampleGa:
,这个平均值可以当作2个标样所夹样品中Ga元素比值的真值,从而可以计算出样品中Ga的分馏系数fsampleGa:
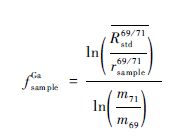
因为fGa≈fCu,所以可以用样品中Ga的分馏系数fsampleCu计算得到样品中Cu同位素比值的真值:

整个校正流程的大体思路是:先在标样中用Cu的分馏系数校正Ga的比值,再在样品中用Ga的分馏系数校正Cu的比值,这种结合法校正所得的数据比单独使用SSB法校正时所得的数据精度提高了5倍左右(Hou et al.,2016),有较广阔的应用前景。
5 不同地质标准物质的测定结果Hou等(2016)首次以Ga作为内标,用内标法与SSB法的结合法(C-SSBIN)校正仪器的质量歧视,并用NIST SRM 3114作为NIST 976的替代标样,对几个常用的国际地质标样(BCR-2,BHVO-2,BIR-1a,AGV-2,GSP-2)进行了测定,根据NIST SRM 3114相对于NIST SRM 976的值(0.18‰±0.04‰),可以将相对于NIST SRM 3114的测定值转换为相对于常用的国际标准物质NIST SRM 976的值,以便与前人的研究数据进行对比。测定结果如 表 1,可以看出,5个岩石标样相对于NIST 976的δ65 Cu在0.01‰~0.26‰,与前人的报道结果一致,进一步表明Hou等(2016)对Cu同位素分析过程中的化学前处理方法、测试方法、标样的选择及校正方法等方面的研究是合理和正确的,为研究Cu同位素提供了新的思路和方法。
|
|
表 1 几种岩石标样的测定值(平均值,2SD) Table 1 Results(mean,2SD)for geological reference materials |
Cu同位素在地质学、气候学、生物学、天文学等研究领域具有广泛的应用前景。近年来,在Cu同位素样品前处理、分离纯化及仪器测定方面的研究取得了诸多进展,可以有效地消除基质元素及多原子元素的干扰,且回收率接近100%,分析精度可以达到0.04‰左右(Takano et al.,2013)。目前,Cu同位素的国际标准物质NIST SRM 976已经停产,对新的替代标准物质的研究尤为迫切,本文讨论了几种替代标样(ERM-AE633、ERM-AE647、GSB Cu、NIST SRM 3114)的研究现状,并提出NIST SRM 3114有可能成为一个新的Cu同位素标准物质。文中介绍了SSB法和内标法(Ga作内标)结合法(C-SSBIN)测定Cu同位素的方法,与传统的直接SSB法相比,C-SSBIN法可以将测试精度提高5倍左右。
| [1] | Albarède F. 2004. The stable isotope geochemistry of copper and zinc. Reviews in Mineralogy and Geochemistry, 55(1): 409-427 |
| [2] | Albarède F, Telouk P, Blichert-Toft J, Boyet M, Agranier A, Nelson B. 2004. Precise and accurate isotopic measurements using multiple-collector ICPMS. Geochimica et Cosmochimica Acta, 68(12): 2725-2744 |
| [3] | Anbar A D, Knab K A, Barling J. 2001. Precise determination of mass-dependent variations in the isotopic composition of molybdenum using MC-ICPMS. Analytical Chemistry, 73(7): 1425-1431 |
| [4] | Archer C, Vance D. 2004. Mass discrimination correction in multiple-collector plasma source mass spectrometry: An example using Cu and Zn isotopes. Journal of Analytical Atomic Spectrometry, 19(5): 656-665 |
| [5] | Baker J, Peate D, Waight T, Meyzen C. 2004. Pb isotopic analysis of standards and samples using a 207Pb-204Pb double spike and thallium to correct for mass bias with a double-focusing MC-ICP-MS. Chemical Geology, 211(3-4): 275-303 |
| [6] | Barbaste M, Robinson K, Guilfoyle S, Medina B, Lobinski R. 2002. Precise determination of the strontium isotope ratios in wine by inductively coupled plasma sector field multicollector mass spectrometry(ICP-SF-MC-MS). Journal of Analytical Atomic Spectrometry, 17(2): 135-137 |
| [7] | Bermin J, Vance D, Archer C, Statham P J. 2006. The determination of the isotopic composition of Cu and Zn in seawater. Chemical Geology, 226(3-4): 280-297 |
| [8] | Bigalke M, Weyer S, Wilcke W. 2010a. Stable copper isotopes: A novel tool to trace copper behavior in hydromorphic soils. Soil Science Society of America Journal, 74(1): 60-73 |
| [9] | Bigalke M, Weyer S, Kobza J, Wilcke W. 2010b. Stable Cu and Zn isotope ratios as tracers of sources and transport of Cu and Zn in contaminated soil. Geochimica et Cosmochimica Acta, 74(23): 6801-6813 |
| [10] | Bigalke M, Weyer S, Wilcke W. 2011. Stable Cu isotope fractionation in soils during oxic weathering and podzolization. Geochimica et Cosmochimica Acta, 75(11): 3119-3134 |
| [11] | Borrok D M, Wanty R B, Ridley W I, Wolf R, Lamothe P J, Adams M. 2007. Separation of copper, iron, and zinc from complex aqueous solutions for isotopic measurement. Chemical Geology, 242(3-4): 400-414 |
| [12] | Brugger J, McPhail D C, Black J, Spiccia L. 2001. Complexation of metal ions in brines: Application of electronic spectroscopy in the study of the Cu(Ⅱ)-LiCl-H2O system between 25 and 90℃. Geochimica et Cosmochimica Acta, 65(16): 2691-2708 |
| [13] | Craddock P R, Rouxel O J, Ball L A, Bach W. 2008. Sulfur isotope measurement of sulfate and sulfide by high-resolution MC-ICP-MS. Chemical Geology, 253(3-4): 102-113 |
| [14] | de Laeter J R, Böhlke J K, De Bièvre P, Hidaka H, Peiser H S, Rosman K J R, Taylor P D P. 2003. Atomic weights of the elements. Review 2000(IUPAC Technical Report). Pure and Applied Chemistry, 75(6): 683-800 |
| [15] | Ehrlich S, Butler I, Halicz L, Rickard D, Oldroyd A, Matthews A. 2004. Experimental study of the copper isotope fractionation between aqueous Cu(Ⅱ) and covellite, CuS. Chemical Geology, 209(3-4): 259-269 |
| [16] | Fletcher I R. 2007. Using the common-Pb standards SRM-981 and SRM-982 as double spikes. International Journal of Mass Spectrometry, 261(2-3): 234-238 |
| [17] | Foster G L, Vance D. 2006. In situ Nd isotopic analysis of geological materials by laser ablation MC-ICP-MS. Journal of Analytical Atomic Spectrometry, 21(3): 288-296 |
| [18] | Gallon C, Aggarwal J, Flegal A R. 2008. Comparison of mass discrimination correction methods and sample introduction systems for the determination of lead isotopic composition using a multicollector inductively coupled plasma mass spectrometer. Analytical Chemistry, 80(22): 8355-8363 |
| [19] | Gélabert A, Pokrovsky O S, Viers J, Schott J, Boudou A, Feurtet-Mazel A. 2006. Interaction between zinc and freshwater and marine diatom species: Surface complexation and Zn isotope fractionation. Geochimica et Cosmochimica Acta, 70(4): 839-857 |
| [20] | Graham S, Pearson N, Jackson S, Griffin W, O'Reilly S Y. 2004. Tracing Cu and Fe from source to porphyry: In situ determination of Cu and Fe isotope ratios in sulfides from the Grasberg Cu-Au deposit. Chemical Geology, 207(3-4): 147-169 |
| [21] | Hou Q H, Zhou L, Gao S, Zhang T, Feng L P, Yang L. 2016. Use of Ga for mass bias correction for the accurate determination of copper isotope ratio in the NIST SRM 3114 Cu standard and geological samples by MC-ICPMS. Journal of Analytical Atomic Spectrometry, 31(1): 280-287 |
| [22] | Ikehata K, Notsu K, Hirata T. 2008. In situ determination of Cu isotope ratios in copper-rich materials by NIR femtosecond LA-MC-ICP-MS. Journal of Analytical Atomic Spectrometry, 23(7): 1003-1008 |
| [23] | Ingle C P, Sharp B L, Horstwood M S A, Parrish R R, Lewis D J. 2003. Instrument response functions, mass bias and matrix effects in isotope ratio measurements and semi-quantitative analysis by single and multi-collector ICP-MS. Journal of Analytical Atomic Spectrometry, 18(3): 219-229 |
| [24] | Kehm K, Hauri E H, Alexander C M O, Carlson R W. 2003. High precision iron isotope measurements of meteoritic material by cold plasma ICP-MS. Geochimica et Cosmochimica Acta, 67(15): 2879-2891 |
| [25] | Korkisch J, Worsfold P J. 1990. Handbook of ion exchange resins: Their application to inorganic analytical chemistry: Volume 1, CRC Press, Boca Raton, FL, 1989(ISBN 0-8493-3191-9). 301 pp. Price £ 100.50. Analytica Chimica Acta, 236: 511 |
| [26] | Kuritani T, Nakamura E. 2003. Highly precise and accurate isotopic analysis of small amounts of Pb using 205Pb-204Pb and 207Pb-204Pb , two double spikes. Journal of Analytical Atomic Spectrometry, 18(12): 1464-1470 |
| [27] | Liu S A, Li D D, Li S G, Teng F Z, Ke S, He Y S, Lu Y H. 2014. High-precision copper and iron isotope analysis of igneous rock standards by MC-ICP-MS. Journal of Analytical Atomic Spectrometry, 29(1): 122-133 |
| [28] | Machlan L A, Gramlich J W, Powell L J, Lambert G M. 1986. Absolute isotopic abundance ratio and atomic weight of a reference sample of gallium. Journal of Research of the National Bureau of Standards, 91(6): 323-331 |
| [29] | Maréchal C N, Télouk P, Albarède F. 1999. Precise analysis of copper and zinc isotopic compositions by plasma-source mass spectrometry. Chemical Geology, 156(1-4): 251-273 |
| [30] | Maréchal C, Albarède F. 2002. Ion-exchange fractionation of copper and zinc isotopes. Geochimica et Cosmochimica Acta, 66(9): 1499-1509 |
| [31] | Mason T F D, Weiss D J, Chapman J B, Wilkinson J J, Tessalina S G, Spiro B, Horstwood M S A, Spratt J, Coles B J. 2005. Zn and Cu isotopic variability in the Alexandrinka volcanic-hosted massive sulphide(VHMS) ore deposit, Urals, Russia. Chemical Geology, 221(3-4): 170-187 |
| [32] | McDonough W F. 2003. Compositional model for the Earth's core. Treatise on Geochemistry, 2: 547-568 |
| [33] | Moeller K, Schoenberg R, Pedersen R B, Weiss D, Dong S F. 2012. Calibration of the new certified reference materials ERM-AE633 and ERM-AE647 for copper and IRMM-3702 for zinc isotope amount ratio determinations. Geostandards and Geoanalytical Research, 36(2): 177-199 |
| [34] | Moynier F, Koeberl C, Beck P, Jourdan F, Telouk P. 2010. Isotopic fractionation of Cu in tektites. Geochimica et Cosmochimica Acta, 74(2): 799-807 |
| [35] | Nowell G M, Pearson D G, Parman S W, Luguet A, Hanski E. 2008. Precise and accurate186Os/188Os and187Os/188Os measurements by multi-collector plasma ionisation mass spectrometry, part Ⅱ: Laser ablation and its application to single-grain Pt-Os and Re-Os geochronology. Chemical Geology, 248(3-4): 394-426 |
| [36] | Pichat S, Douchet C, Albarède F. 2003. Zinc isotope variations in deep-sea carbonates from the eastern equatorial Pacific over the last 175 ka. Earth and Planetary Science Letters, 210(1-2): 167-178 |
| [37] | Pribil M J, Wanty R B, Ridley W I, Borrok D M. 2010. Influence of sulfur-bearing polyatomic species on high precision measurements of Cu isotopic composition. Chemical Geology, 272(1-4): 49-54 |
| [38] | Ramette R W. 1986. Copper(Ⅱ) complexes with chloride ion. Inorganic Chemistry, 25(14): 2481-2482 |
| [39] | Rudnick R L, Gao S. 2003. Composition of the continental crust. Treatise on Geochemistry, 3: 1-64 |
| [40] | Russell W A, Papanastassiou D A, Tombrello T A. 1978. Ca isotope fractionation on the Earth and other solar system materials. Geochimica et Cosmochimica Acta, 42(8): 1075-1090 |
| [41] | Saito M A, Schneider D L. 2006. Examination of precipitation chemistry and improvements in precision using the Mg(OH)2 preconcen ̄tration inductively coupled plasma mass spectrometry(ICP-MS) method for high-throughput analysis of open-ocean Fe and Mn in seawater. Analytica Chimica Acta, 565(2): 222-233 |
| [42] | Shields W R, Goldich S S, Garner E L, Murphy T J. 1965. Natural variations in the abundance ratio and the atomic weight of copper. Journal of Geophysical Research, 70(2): 479-491 |
| [43] | Takano S, Tanimizu M, Hirata T, Sohrin Y. 2013. Determination of isotopic composition of dissolved copper in seawater by multi-collector inductively coupled plasma mass spectrometry after pre-concentration using an ethylenediaminetriacetic acid chelating resin. Analytica Chimica Acta, 784: 33-41 |
| [44] | Taylor S R, McLennan S M. 1985. The continental crust: Its composition and evolution. Oxford: Blackwell Scientific Publications |
| [45] | Thirlwall M F. 2000. Inter-laboratory and other errors in Pb isotope analyses investigated using a 207Pb-204Pb double spike. Chemical Geology, 163(1-4): 299-322 |
| [46] | Thompson C M, Ellwood M J. 2014. Dissolved copper isotope biogeochemistry in the Tasman Sea, SW Pacific Ocean. Marine Chemistry, 165: 1-9 |
| [47] | van der Walt T N, Strelow F W E, Verheij R. 1985. The influence of crosslinkage on the distribution coefficients and anion exchange behaviour of some elements in hydrochloric acid. Solvent Extraction and Ion Exchange, 3(5): 723-740 |
| [48] | Vance D, Archer C, Bermin J, Perkins J, Statham P J, Lohan M C, Ellwood M J, Mills R A. 2008. The copper isotope geochemistry of rivers and the oceans. Earth and Planetary Science Letters, 274(1-2): 204-213 |
| [49] | Waight T, Baker J, Willigers B. 2002. Rb isotope dilution analyses by MC-ICPMS using Zr to correct for mass fractionation: Towards improved Rb-Sr geochronology? Chemical Geology, 186(1-2): 99-116 |
| [50] | Walker E C, Cuttitta F, Senftle F E. 1958. Some natural variations in the relative abundance of copper isotopes. Geochimica et Cosmochimica Acta, 15(3): 183-194 |
| [51] | Weinstein C, Moynier F, Wang K, Paniello R, Foriel J, Catalano J. 2011. Isotopic fractionation of Cu in plants. Chemical Geology, 286(3-4): 266-271 |
| [52] | Wu J F, Boyle E A. 1997. Low blank preconcentration technique for the determination of lead, copper, and cadmium in small-volume seawater samples by isotope dilution ICPMS. Analytical Chemistry, 69(13): 2464-2470 |
| [53] | Xie Q L, Lu S Y, Evans D, Dillon P, Hintelmann H. 2005. High precision Hg isotope analysis of environmental samples using gold trap-MC-ICP-MS. Journal of Analytical Atomic Spectrometry, 20(6): 515-522 |
| [54] | Yang L. 2009. Accurate and precise determination of isotopic ratios by MC-ICP-MS: A review. Mass Spectrometry Reviews, 28(6): 990-1011 |
| [55] | Yang L, Sturgeon R E. 2009. Isotopic fractionation of mercury induced by reduction and ethylation. Analytical and Bioanalytical Chemistry, 393(1): 377-385 |
| [56] | Yang L, Meija J. 2010. Resolving the germanium atomic weight disparity using multicollector ICPMS. Analytical Chemistry, 82(10): 4188-4193 |
| [57] | Yang L, Sturgeon R E, Mester Z, Meija J. 2010. Metrological triangle for measurements of isotope amount ratios of silver, indium, and antimony using multicollector-inductively coupled plasma mass spectrometry: The 21 st century harvard method. Analytical Chemistry, 82(21): 8978-8982 |
| [58] | Zheng W, Foucher D, Hintelmann H. 2007. Mercury isotope fractionation during volatilization of Hg(0) from solution into the gas phase. Journal of Analytical Atomic Spectrometry, 22(9): 1097-1104 |
| [59] | Zhou L, Huang J H, Archer C, Hawkesworth C. 2007. Molybdenum isotope composition from Yangtze block continental margin and its indication to organic burial rate. Frontiers of Earth Science in China, 1(4): 417-424 |
| [60] | Zhou L, Su J, Huang J H, Yan J X, Xie X N, Gao S, Dai M N. 2011. A new paleoenvironmental index for anoxic events-Mo isotopes in black shales from Upper Yangtze marine sediments. Science China Earth Sciences, 54(7): 1024-1033 |
| [61] | Zhou L, Wignall P B, Su J, Feng Q L, Xie S C, Zhao L S, Huang J H. 2012. U/Mo ratios and δ98/95Mo as local and global redox proxies during mass extinction events. Chemical Geology, 324-325: 99-107 |
| [62] | Zhu X K, O'Nions R K, Guo Y, Belshaw N S, Rickard D. 2000. Determination of natural Cu-isotope variation by plasma-source mass spectrometry: Implications for use as geochemical tracers. Chemical Geology, 163(1-4): 139-149 |
| [63] | 蔡俊军, 朱祥坤, 唐索寒, 李世珍, 何学贤. 2006. 多接收电感耦合等离子体质谱Cu同位素测定中的干扰评估. 高校地质学报, 12(3): 392-397 |
| [64] | 侯可军, 李延河, 田有荣, 秦燕, 谢桂青. 2008. MC-ICP-MS高精度Cu、Zn同位素测试技术. 矿床地质, 27(6): 774-781 |
| [65] | 蒋少涌, Woodhead J, 于际民, 潘家永, 廖启林, 吴南平. 2001. 云南金满热液脉状铜矿床 Cu 同位素组成的初步测定. 科学通报, 46(17): 1468-1471 |
| [66] | 李丹丹. 2015. 低温过程中Cu-Zn同位素分馏的实验地球化学研究. 博士学位论文. 北京: 中国地质大学(北京) |
| [67] | 李世珍, 朱祥坤, 吴龙华, 骆永明. 2011. 干法灰化和湿法消解植物样品的铜锌铁同位素测定对比研究. 地球学报, 32(6): 754-760 |
| [68] | 梁莉莉, 刘丛强, 王中良, 宋柳霆. 2006. 铜锌同位素方法在环境地球化学研究中的应用. 地球与环境, 34(1): 81-89 |
| [69] | 马信江, 梁细荣, 涂相林, 曾文, 李杰. 2009. AG MP-1M阴离子分离Cu、Fe、Zn及其在Fe同位素测定上的应用. 地球化学, 38(5): 480-486 |
| [70] | 孙剑, 朱祥坤, 唐索寒, 陈岳龙. 2009. AGMP-1阴离子交换树脂元素分离方法再研究. 矿物学报,(S): 613-614 |
| [71] | 唐索寒, 朱祥坤. 2006. AG MP-1阴离子交换树脂元素分离方法研究. 高校地质学报, 12(3): 398-403 |
| [72] | 唐索寒, 朱祥坤, 蔡俊军, 李世珍, 何学贤, 王进辉. 2006. 用于多接收器等离子体质谱铜铁锌同位素测定的离子交换分离方法. 岩矿测试, 25(1): 5-8 |
| [73] | 唐索寒, 闫斌, 朱祥坤, 李津, 李世珍. 2012. 玄武岩标准样品铁铜锌同位素组成. 岩矿测试, 31(2):218-224 |
| [74] | 王家松, 彭丽娜. 2012. 铜同位素样品化学前处理方法的研究进展. 理化检验-化学分册, 48(11): 1383-1388 |
| [75] | 王跃, 朱祥坤. 2010. 铜同位素在矿床学中的应用: 认识与进展. 吉林大学学报(地球科学版), 40(4): 739-751 |
| [76] | 赵振华. 1992. 微量元素地球化学. 地球科学进展, 7(5): 65-66 |