 2016, Vol. 35
2016, Vol. 35



2. 中国地质科学院 地质研究所, 北京离子探针中心, 北京 102206;
3. 南京大学 中国南海研究协同创新中心, 南京 210093
2. Beijing SHRIMP Center, Institute of Geology, Chinese Academy of Geological Sciences, Beijing 102206, China;
3. Collaborative Innovation Center of South China Sea Studies, Nanjing University, Nanjing 210093, China
硫是一种相对活泼的元素,它有4种稳定同位素(32S、33S、34S和36S),其相对丰度分别是94.93%、0.76%、4.29%和0.02%(Rosman and Taylor,1998)。在自然界中,硫有多种氧化态,常见的赋存形式包括硫酸盐,单质硫和硫化物。这些氧化态的变化主要来自于生物过程和地球化学过程,并且这些过程会引起硫同位素的分馏,导致自然界中硫同位素组成(δ34 S)的变化范围很大,从-65‰到+90‰(Thode et al.,1961)。也由于此,地质样品中的硫同位素组成可以有效示踪生物地球化学过程中硫的来源和循环过程(Thode et al.,1961; Canfield,2001; Shanks Ⅲ,2001)。在硫同位素的实际应用中,利用34S/32S就能够示踪硫的来源和研究其分馏过程。本文将主要介绍 δ34 S 的测定方法。
传统的硫同位素组成分析方法是气源质谱法(GS-MS)。对气源质谱而言,硫是以气态的SO2或SF6引入的(Thode et al.,1961; Fritz et al.,1974; Robinson and Kusakabe,1975; Rees,1978)。SF6法通常用于高精度的硫同位素分析,因为氟只有一种同位素,不会对测试造成叠加干扰(Rees,1978)。但是SF6的制备过程繁琐且危险,这限制了该方法的广泛使用。SO2法相对于SF6法而言较为安全,但是样品的前处理过程也较为复杂,并且SO2的记忆效应严重,容易吸附在管道上。连续流质谱法(CF-MS)也已经被广泛用于硫同位素的分析,在用连续流质谱进行硫同位素测定时,硫以SO2的形式进入质谱进行分析,并且能实现SO2制备和纯化过程的自动化(Giesemann et al.,1994; Grassineau et al.,2001; Studley et al.,2002)。因此与气源质谱法相比,分析速度得到了提升。但是,自动化的样品制备系统不能很好地约束氧同位素组成的变化,从而导致最终得到的 δ34 S 值存在高达1‰~3‰的误差(Fry et al.,2002)。
硫同位素的测定还有一些其他的方法,例如多接收热电离质谱法(MC-TIMS)和二次离子质谱法(SIMS)。热电离质谱是以AsS+的形式进行硫同位素测定的,并且测试所需的样品量很少(<2 μg S)(Paulsen and Kelly,1984)。但是样品的制备过程繁琐而费时。二次离子质谱法能进行硫同位素的原位分析,并且具有很高的空间分辨率(8~25 μm)。但测试过程中的谱学干扰和基体效应较为严重,并且只能进行原位测量,而不适用于溶液样品。
近20年来,多接收电感耦合等离子体质谱(MC-ICP-MS)技术发展迅速,它具有进样简单高效,样品测试效率高和分辨率高等优点,并且能与多种进样方式联用,同时满足溶液测试和固体原位测量的需求(Halliday et al.,1998; Rehkämper et al.,2001; Mason et al.,2006)。此外,MC-ICP-MS的平顶峰也保证了它测试过程中的准度和精度(精度可达0.001%)(Albarède et al.,2004)。MC-ICP-MS的上述特征,再加上ICP离子源强大的电离能力(几乎可以电离元素周期表中所有元素),使得MC-ICP-MS的应用日益广泛,并且大大推动了稳定同位素(如锂、硼、硫、铁同位素等)的发展(Aggarwal et al.,2003; Jeffcoate et al.,2004; Craddock et al.,2008; 朱祥坤等,2008)。
Clough等(2006)率先建立了硫同位素的MC-ICP-MS测定方法,该方法是向含硫溶液中加入硅标准溶液并通过30Si/28Si同位素对来校正硫的质量歧视。但这一方法无法保证30Si/28Si同位素对和硫同位素的质量歧视校正因子完全一致,因而在之后的研究中更多的是使用样品-标样交叉法(SSB法)来进行仪器的质量歧视校正(Craddock et al.,2008; Lin et al.,2014; Bian et al.,2015; Hanousek et al.,2016)。Craddock等(2008)建立了更全面的硫同位素MC-ICP-MS测定方法,涉及的范围包括了地质样品(硫酸盐和硫化物)的溶解和提纯,仪器的质量歧视校正,谱学干扰的避免和基体效应的评价和校正。除此之外,对溶液进样和激光剥蚀进样这2种进样方式都进行了尝试。之后,硫同位素的MC-ICP-MS测定方法发展更为迅速,对MC-ICP-MS硫同位素分析的各个方面都有了更深入的研究。值得一提的是,早期硫同位素测试主要是通过对样品进行化学提纯来避免基体效应。但最近基体匹配方法也广泛用于硫同位素测试过程中的基体效应校正(Lin et al.,2014; Bian et al.,2015; Liu et al.,2016)。
尽管硫同位素的MC-ICP-MS测定方法已经建立和发展了近十年,但依旧有许多值得关注和解决的问题,主要包括谱学干扰的避免、对基体效应的评估和校正及对浓度效应的探究。本文将对硫同位素的MC-ICP-MS测定方法进行详细的介绍,并对上述问题进行进一步的阐述。
1 MC-ICP-MS简介目前最常用的MC-ICP-MS主要包括Nu Instruments公司生产的Nu Plasma HR和ThermoFisher Scientific公司生产的Neptune/Neptune Plus。Nu Plasma HR型MC-ICP-MS配有12个法拉第杯检测器;Neptune Plus型MC-ICP-MS配有9个法拉第杯检测器,除中心杯固定外,中心杯两侧的8个法拉第杯都可以移动。Nu Plasma HR和Neptune Plus型MC-ICP-MS都具有Nier-Johnson几何结构,并且都是双聚焦的扇形磁场质谱仪(Yang,2009)。Nu Plasma HR和Neptune Plus型MC-ICP-MS具有相似的灵敏度,它们的测试精度都可以达到0.001%~0.002%。此外,它们都能够通过降低入口狭缝的宽度在中-高分辨率模式下进行测试,从而将待测同位素和它的谱峰干扰区分开。最典型的例子是在Fe同位素测试时,在中-高分辨率模式下,能够将Fe和它的谱峰干扰(例如 40 Ar16O对56Fe的干扰)区分开,从而避免谱学干扰。本文接下来的报道主要是基于Neptune Plus型MC-ICP-MS的测试过程和分析结果。硫同位素在测试中需要将分辨率模式调节至中分辨率,从而避免来自双氧原子团的干扰(图 1)。关于硫的谱学干扰在2.5节会有更详细的说明。

|
图 1 硫同位素测试过程中的谱学干扰 Fig. 1 pectral interferences on signals of sulfur isotopes |
在硫同位素的MC-ICP-MS测试过程中,在样品前后需进行标样的测试。对于标样的选择通常有2种:一种是使用硫标准物质IAEA-S-1,S-2,它是人工合成的Ag2S固体,将其消解纯化后即可作为标样使用(Craddock et al.,2008; Hanousek et al.,2016);另一种是用硫酸铵溶液或溶解的硫酸铵固体作为标准(Craddock et al.,2008; Lin et al.,2014; Bian et al.,2015; Hanousek et al.,2016)。作者所在的南京大学内生金属矿床成矿机制研究国家重点实验室(以下简称实验室)通常采用硫酸铵溶液作为测试时的标样。常见的国际标样和实验室所用标样的性质和硫同位素值(δ34 SV-CDT值)见 表 1。
|
|
表 1 国际标样和实验室所用标样的性质和硫同位素值 Table 1 values of international and laboratory reference materials |
在硫同位素测试过程中,为了得到足够的精度,通常在10 mg/L的硫浓度下(约3 V的信号)进行测试(详见3.3节)。
2.2 样品消解与纯化MC-ICP-MS最常见的进样方式是溶液进样,因此,在测试前需要对固体样品进行消解。Craddock等(2008)详细描述了对硫化物的消解过程,所有的硫最终都被转化为硫酸根,在此不再赘述。对于可溶解的硫酸盐(如石膏),可直接在18.2 MΩ cm的Milli-Q去离子水中加热溶解即可。
因为在硫同位素测试时需要用SSB法进行质量歧视校正,而SSB法成功的前提是保证样品和标样的性质一致(Albarède et al.,2004),即基体和浓度一致(详见第3节)。由于硫同位素分析中作为标准的物质(如硫酸铵溶液和消解提纯过后的Ag2S固体)往往不含有其他的基体,因此,在样品消解后,还需要提纯。通常硫化物和硫酸盐,在消解之后,硫以硫酸根的形式存在于溶液中,而基体主要是阳离子,因此只需要用阳离子交换树脂(如AG50-X8)进行纯化即可(Craddock et al.,2008)。对于基体较为复杂的样品,如沉积物孔隙水和土壤中的水样(硫以硫酸根形式存在),除了阳离子外,阴离子的成分也较为复杂,可以使用阴离子交换膜(如VWR公司的551642S型阴离子交换膜)来进行提纯(Kwon et al.,2008; Hanousek et al.,2016)。
2.3 样品进样方式本文所介绍的硫同位素的MC-ICP-MS测定方法主要基于溶液进样。而溶液进样方式又可细分为2种:①湿法进样,即用常规的PFA雾化器和石英雾化室进行进样;②干法进样,是将溶液雾化之后再通过膜去溶装置(例如Aridus Ⅱ型膜去溶装置)进入等离子体。干法进样能够提高仪器的灵敏度,还可以较大幅度地减少等离子体中H2O、N2、CO2等的进入量,进而降低一些潜在的谱学干扰,但在有基体存在的情况下,干法进样会引起更明显的基体效应(Liu et al.,2016)。
2.4 质量歧视校正和数据处理硫同位素测试是通过SSB法进行仪器的质量歧视校正。硫同位素比值的测定结果以样品相对于国际标准(V-CDT)的千分偏差表示(Krouse and Coplen,1997):
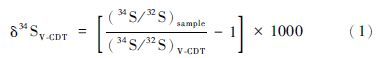
实际测试中,为了得到样品的δ34 SV-CDT值,往往用一种已知δ34 SV-CDT值的物质作为标准(表 1),并且由于是使用SSB法进行测量,因此样品的δ34 SV-CDT值可直接由下式求得:
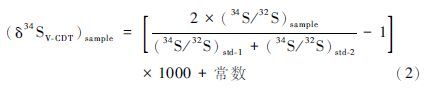
同位素测试过程中的谱学干扰(spectral interferences)主要包括同质异位素干扰(isobaric interferences),二价离子干扰和多原子离子干扰。在硫同位素测试过程中,谱学干扰主要来自氧的双原子团(如16O16O、16O17O和16O18O),通过切换MC-ICP-MS的分辨率模式,在中分辨率模式(m/Δm≈3000)下,就可以出现一个没有干扰的平台,将硫同位素与其谱学干扰区分开(表 2,图 1)。这里仪器分辨率的计算是依据传统的Rpower(5,95%)公式(Weyer and Schwieters,2003)(m代表峰位置处的质量数,Δm代表 5%峰高处和95%峰高处的质量差)。由于36S的天然丰度很低,不具有地质意义,因此实验中未予测定,相应地,表 2中也没有列出可能影响36S测试的谱学干扰。在实际测试时,只需要在没有谱学干扰的平台处(图 1实线处)进行测量就能得到准确的分析结果(图 1)。
|
|
表 2 硫同位素测试过程中一些潜在的谱学干扰 Table 2 Some potential spectral interferences on signals of sulfur isotopes |
如上所述,硫同位素测试过程中是通过SSB法校正仪器的质量歧视。实际上,SSB法被广泛用于稳定同位素的质量歧视校正中(Albarède et al.,2004)。其前提假设是:在样品和标样性质一样的情况下,测试过程中样品和标样的质量歧视随时间有同样的响应。基于此,就能通过样品前后的标样来校正样品的质量歧视。所以,测试过程中,保证样品和标样具有相同的性质至关重要。样品和标样的性质一致主要包括基体和浓度的一致。
3.1 基体效应基体效应是质谱分析中的共性问题,待测样品中除了待测元素外,往往还含有其他物质,其他物质相对于测定元素而言就是基体。基体的存在对同位素测试主要有2方面的影响:一是可能引起更多的谱学干扰,二是在基体差异大的情况下会使样品和标样的质量歧视出现明显变化,从而使运用SSB法校正后的测量值偏离真实值。后者也就是通常所说的基体效应(matrix effects)。在硫同位素测试过程中,硫的谱学干扰主要来源于双氧原子团,并且在中-高分辨率模式下能得到很好的区分和扣除。所以对硫同位素测定而言,基体对同位素测试的影响主要表现在影响仪器的质量歧视上。
3.1.1基体对硫同位素值测定的影响MC-ICP-MS测试时的仪器质量歧视通常归因于离子在传输过程中的扩散效应(ion-diffusion effects)和空间电荷效应(space-charge effects)(Tanner,1992; Vanhaecke et al.,1993),并且仪器的质量歧视对基体十分敏感,这是基体效应产生的原因。为了避免基体效应,需要保证样品和标样的基体一致。由于标样通常不含除待测元素外的其他基体,因此对样品进行纯化是避免基体效应的一种十分有效的手段。另一种校正基体效应的方法是基体匹配,即通过向标样中加入适当的基体,使得标样和样品的性质一致(Lin et al.,2014; Bian et al.,2015)。但无论何种方法,都需要对基体效应进行研究。一方面,只有当基体的存在显著影响测试值时,上述的2种校正基体效应的方法才有应用的必要;另一方面,了解基体的存在是如何影响测试值的,也有助于更好地消除基体的影响。
海水是地球上硫酸根的最大储库之一,而NaCl是海水中最主要的基体,因此首先对NaCl在硫同位素测试过程中的基体效应进行评估。实验过程是向10 mg/L的Alfa-S(AS)溶液(表 1)中加入不同浓度的NaCl,并用纯的10 mg/L的AS溶液作为标样进行测试。如 图 2所示,尽管AS溶液拥有相同的硫同位素组成,但随着NaCl浓度的增加,测试值相对于真实值的偏差逐渐加大。最大的偏差出现在Cl-浓度为800 mg/L时,偏差达到-2.19‰。 图 2中△34S表示测量值与真实值之间的差值,具体而言,△34S=(δ34 SV-CDT)测量值-(δ34 SV-CDT)真实值,图中的虚线表示△34S为0的情况,即测量值等于真实值。由于NaCl基体会对硫同位素测量值造成显著的影响,因此需要对此进行校正。通过基体匹配的方法,向AS溶液中加入等量的NaCl,就能消除上述NaCl造成的基体效应。这种基体匹配的硫酸铵溶液也成功被用于测试海水和孔隙水中的硫同位素(Lin et al.,2014)。

|
图 2 NaCl在硫同位素测试过程中的基体效应(据Lin等,2014修改) Fig. 2 atrix effects of NaCl on sulfur isotopes during the measurement |
除了上述对NaCl基体的研究之外,许多研究者也对其他的阴阳离子(例如Li+、K+、Ca2+、Mg2+、Zn2+和PO3-4等)在硫同位素测试过程中的基体效应做了研究(Craddock et al.,2008; Hanousek et al.,2016)。但是这些研究更多地是关注基体效应是否存在,以此来证明化学提纯或基体匹配的必要性,而对基体效应的显著程度是由基体的那种属性引发的则关注较少,甚至给出了一些模棱两可的结论。例如Hanousek等(2016)认为基体/待测元素的相对浓度比值会显著影响基体效应的程度。但是他们的结论是基于单一硫浓度条件下的基体效应研究结果。
基于此,Liu等(2016)以Ca2+基体为研究对象,对基体效应作了更为深入的研究。选择Ca2+作为研究对象是因为它是一种重要的蒸发岩矿物-石膏的主要基体。在实验过程中,配置了2组不同浓度的AS溶液(0.3 mM和0.6 mM),对每个特定的S浓度,都配置了7组特定Ca/S摩尔比的溶液(Ca/S摩尔比分别是0.1、0.4、0.7、1、5、10和25)。对每组特定浓度的AS溶液,都以纯的AS溶液作为标样来测试含基体的AS溶液。测量值与真实值之间的差值依旧用△34S表示。如 图 3(a)所示,当以Ca/S摩尔比为横坐标时,随着Ca/S值的增加,2组不同浓度的AS溶液呈现出相似的变化趋势,但2条曲线并不重叠。在同一Ca/S值的条件下,高浓度组的AS溶液表现出更明显的基体效应,即△34S的绝对值更大。为了判断影响基体效应显著程度的关键因素是基体/待测元素的相对浓度比还是基体的绝对浓度,Ca的绝对浓度也被当作横坐标作了类似的图。如 图 3(b)所示,与 图 3(a)不同,所有的点都落在了相同的趋势线上,随着Ca的绝对浓度的增加,Δ34S的绝对值也逐渐变大。这强有力地反映了Ca的绝对浓度是影响基体效应显著程度的关键。

|
图 3 Ca基体对硫同位素测试的影响(据Liu等(2016)修改) Fig. 3 The effects of calcium matrix on sulfur isotope ratios(revised from Liu et al.,2016) |
基体的存在除了会影响同位素比值外,还会影响待测元素的灵敏度。一般基体的存在会引起信号的抑制,主要是由于空间电荷效应(Tanner,1992)。但少量盐类基体的存在会诱发库伦裂变(Coulomb fission),使得在雾化过程中产生更小的液滴,进而引起信号的增益(Xu et al.,2001; Fraser and Beauchemin,2009)。
因此,利用3.1.1节中的2组不同浓度的AS溶液,Liu等(2016)探究了基体对硫信号值的影响。如 图 4(a)和图 4(b)所示,纵坐标都为I样品/I标样,表示的是样品的32S信号与样品前后2个标样的32S信号平均值的比值,因此,当I样品/I标样大于1时,表示信号增益;而当I样品/I标样小于1时,表示信号受到了抑制。当以Ca/S摩尔比为横坐标时,随着Ca/S比的增加,2组不同浓度的AS溶液都呈现出相似的变化趋势,即信号先增高,后抑制。但2条曲线并不重叠;当以Ca的绝对浓度为横坐标时,类似地,所有的点都落在了相同的趋势线上,随着Ca的绝对浓度的增加,样品的信号呈现出先增高后抑制的趋势。这也反映了基体对硫信号值的影响也主要受控于基体的绝对浓度。而样品信号的这种先增益后抑制的现象应是库伦裂变和空间电荷效应共同作用的结果。

|
图 4 Ca基体对硫信号的影响(据Liu等,2016修改) Fig. 4 The effects of calcium matrix on sulfur signals(revised from Liu et al.,2016) |
前文提到,样品和标样的性质一致包括了基体和浓度的一致。关于基体效应前文中已有详细论述,这里介绍一下硫同位素测试过程中的浓度效应。所谓浓度效应,是指在测试过程中,由于样品和标样浓度的差异所造成的测量值与真实值之间的偏差。关于浓度效应,Bian等(2015)做了细致的研究。实验过程中,分别配制了从1 mg/L到20 mg/L的一系列不同浓度的AS溶液,并以10 mg/L的AS溶液作为标样进行测试。如 图 5所示,当样品的信号低于标样时,所测得的硫同位素值低于真实值(最高可低-25.55‰);当样品的信号高于标样时,所测得的硫同位素值高于真实值。并且这种定量化的描述可以被定性表达,即I标样/I样品和△34S呈线性关系,其中I标样/I样品和△34S的含义与3.1节中的相同。利用这种线性关系,可以成功地校正硫的浓度效应(Bian et al.,2015)。因此,在大批量测试硫浓度变化范围较大的样品时,不需要进行浓度匹配,利用该浓度效应校正曲线能使测试变得更为简便快捷。

|
图 5 硫同位素测试过程中的浓度效应(据Bian等,2015修改) Fig. 5 oncentration effects on sulfur isotopic ratios during the measurement(revised from Bian et al.,2015) |
在硫同位素测试过程中,为了得到足够的精度,通常在10 mg/L的硫浓度下(约3 V的信号)进行测试。测试结果的内部精度和外部精度见 图 6。从 图 6可以看出,内部精度均优于0.15‰(2SE),10次测试所得到的外部精度为0.17‰(2SD)。上述的数据质量能完全满足硫同位素的测试需求,并且在仪器正常的情况下均能达到。

|
图 6 硫同位素测试的长期稳定性实验 Fig. 6 Long-term repeatability of sulfur isotope measurement over multiple, independent analytical sessions |
运用所建方法,笔者对孔隙水和石膏中的硫同位素组成进行了分析。其中孔隙水的硫同位素测试是采用基体匹配的硫酸铵溶液作为标样,而石膏的硫同位素分析是采用纯的硫酸铵溶液作为标样(因为Ca绝对浓度很低时,基体效应不明显)。由其分析结果(表 3)可见,所有数据都与MAT253稳定同位素比值质谱仪给出的参考值一致,再次验证了所建方法的可靠性。
|
|
表 3 孔隙水和石膏样品的硫同位素组成 Table 3 Sulfur isotopic compositions of pore water and gypsum samples |
(1)在硫同位素测试过程中,可以利用MC-ICP-MS的高分辨率避免测试中的谱学干扰,可用SSB法进行仪器的质量歧视校正。在利用SSB法进行测试时,样品与标样的基体要一致,浓度要匹配。
(2)基体的存在既能影响硫同位素值的测定,也能影响硫的信号。但这些影响主要受控于基体的绝对浓度。为了避免基体效应,可以对样品进行化学提纯或配制基体匹配的标样。
(3)硫同位素测试过程中浓度效应十分显著,但利用浓度效应校正曲线可以有效校正浓度效应,并使测试更为简便快捷。
(4)MC-ICP-MS硫同位素测试过程中的内部精度和外部精度都完全能满足应用的需求。
| [1] | Aggarwal J K, Sheppard D, Mezger K, Pernicka E. 2003. Precise and accurate determination of boron isotope ratios by multiple collector ICP-MS: Origin of boron in the Ngawha geothermal system, New Zealand. Chemical Geology, 199(3-4): 331-342 |
| [2] | Albarède F, Telouk P, Blichert-Toft J, Boyet M, Agranier A, Nelson B. 2004. Precise and accurate isotopic measurements using multiple-collector ICPMS. Geochimica et Cosmochimica Acta, 68(12): 2725-2744 |
| [3] | Bian X P, Yang T, Lin A J, Jiang S Y. 2015. Rapid and high-precision measurement of sulfur isotope and sulfur concentration in sediment pore water by multi-collector inductively coupled plasma mass spectrometry. Talanta, 132: 8-14 |
| [4] | Canfield D E. 2001. Biogeochemistry of sulfur isotopes. Reviews in Mineralogy and Geochemistry, 43(1): 607-636 |
| [5] | Clough R, Evans P, Catterick T, Evans E H. 2006. δ34S measurements of sulfur by multicollector inductively coupled plasma mass spectrometry. Analytical Chemistry, 78(17): 6126-6132 |
| [6] | Coplen T B, Krouse H R. 1998. Sulphur isotope data consistency improved. Nature, 392(6671): 32 |
| [7] | Craddock P R, Rouxel O J, Ball L A, Bach W. 2008. Sulfur isotope measurement of sulfate and sulfide by high-resolution MC-ICP-MS. Chemical Geology, 253(3-4): 102-113 |
| [8] | Ding T, Valkiers S, Kipphardt H, De Bièvre P, Taylor P D P, Gonfiantini R, Krouse R. 2001. Calibrated sulfur isotope abundance ratios of three IAEA sulfur isotope reference materials and V-CDT with a reassessment of the atomic weight of sulfur. Geochimica et Cosmochimica Acta, 65(15): 2433-2437 |
| [9] | Fraser M M, Beauchemin D. 2009. Evidence supporting the occurrence of Coulomb fission during conventional sample introduction in inductively coupled plasma mass spectrometry. Journal of Analytical Atomic Spectrometry, 24(4): 469-475 |
| [10] | Fritz P, Drimmie R J, Nowicki V K. 1974. Preparation of sulfur dioxide for mass spectrometer analyses by combustion of sulfides with copper oxide. Analytical Chemistry, 46(1): 164-166 |
| [11] | Fry B, Silva S R, Kendall C, Anderson R K. 2002. Oxygen isotope corrections for online δ34S analysis. Rapid Communications in Mass Spectrometry, 16(9): 854-858 |
| [12] | Giesemann A, Jäger H J, Norman A L, Krouse H R, Brand W A. 1994. Online sulfur-isotope determination using an elemental analyzer coupled to a mass spectrometer. Analytical Chemistry, 66(18): 2816-2819 |
| [13] | Grassineau N V, Mattey D P, Lowry D. 2001. Sulfur isotope analysis of sulfide and sulfate minerals by continuous flow-isotope ratio mass spectrometry. Analytical Chemistry, 73(2): 220-225 |
| [14] | Halliday A N, Lee D C, Christensen J N, Rehkämper M, Yi W, Luo X Z, Hall C M, Ballentine C J, Pettke T, Stirling C. 1998. Applications of multiple collector-ICPMS to cosmochemistry, geochemistry, and paleoceanography. Geochimica et Cosmochimica Acta, 62(6): 919-940 |
| [15] | Hanousek O, Berger T W, Prohaska T. 2016. MC ICP-MS δ34SVCDT measurement of dissolved sulfate in environmental aqueous samples after matrix separation by means of an anion exchange membrane. Analytical and Bioanalytical Chemistry, 408(2): 399-407 |
| [16] | Jeffcoate A B, Elliott T, Thomas A, Bouman C. 2004. Precise/small sample size determinations of lithium isotopic compositions of geological reference materials and modern seawater by MC-ICP-MS. Geostandards and Geoanalytical Research, 28(1): 161-172 |
| [17] | Krouse H R, Coplen T B. 1997. Reporting of relative sulfur isotope-ratio data(technical report). Pure and Applied Chemistry, 69(2): 293-296 |
| [18] | Kwon J S, Mayer B, Yun S T, Nightingale M. 2008. The use of ion exchange membranes for isotope analyses on soil water sulfate: Laboratory experiments. Journal of Environmental Quality, 37(2): 501-508 |
| [19] | Lin A J, Yang T, Jiang S Y. 2014. A rapid and high-precision method for sulfur isotope δ34S determination with a multiple-collector inductively coupled plasma mass spectrometer: Matrix effect correction and applications for water samples without chemical purification. Rapid Communications in Mass Spectrometry, 28(7): 750-756 |
| [20] | Liu C H, Bian X P, Yang T, Lin A J, Jiang S Y. 2016. Matrix effects of calcium on high-precision sulfur isotope measurement by multiple-collector inductively coupled plasma mass spectrometry. Talanta, 151: 132-140 |
| [21] | Mason P R D, Košler J, de Hoog J C M, Sylvester P J, Meffan-Main S. 2006. In situ determination of sulfur isotopes in sulfur-rich materials by laser ablation multiple-collector inductively coupled plasma mass spectrometry(LA-MC-ICP-MS). Journal of Analytical Atomic Spectrometry, 21(2): 177-186 |
| [22] | Paulsen P J, Kelly W R. 1984. Determination of sulfur as arsenic monosulfide ion by isotope dilution thermal ionization mass spectrometry. Analytical Chemistry, 56(4): 708-713 |
| [23] | Rees C E. 1978. Sulphur isotope measurements using SO2 and SF6. Geochimica et Cosmochimica Acta, 42(4): 383-389 |
| [24] | Rehkämper M, Schönbächler M, Stirling C H. 2001. Multiple collector ICP-MS: Introduction to instrumentation, measurement techniques and analytical capabilities. Geostandards Newsletter, 25(1): 23-40 |
| [25] | Robinson B W, Kusakabe M. 1975. Quantitative preparation of sulfur dioxide, for sulfur-34/sulfur-32 analyses, from sulfides by combustion with cuprous oxide. Analytical Chemistry, 47(7): 1179-1181 |
| [26] | Rosman K J R, Taylor P D P. 1998. Isotopic compositions of the elements 1997(Technical Report). Pure and Applied Chemistry, 70(1): 217-235 |
| [27] | Shanks Ⅲ W C. 2001. Stable isotopes in seafloor hydrothermal systems: Vent fluids, hydrothermal deposits, hydrothermal alteration, and microbial processes. Reviews in Mineralogy and Geochemistry, 43(1): 469-525 |
| [28] | Studley S A, Ripley E M, Elswick E R, Dorais M J, Fong J, Finkelstein D, Pratt L M. 2002. Analysis of sulfides in whole rock matrices by elemental analyzer-continuous flow isotope ratio mass spectrometry. Chemical Geology, 192(1-2): 141-148 |
| [29] | Tanner S D. 1992. Space charge in ICP-MS: Calculation and implications. Spectrochimica Acta Part B: Atomic Spectroscopy, 47(6): 809-823 |
| [30] | Taylor B, Ding T, Halas S, Breas O, Robinson B. 2000. Accurate calibration of the V-CDT scale: Proposed δ34S values for calibration and reference materials and methods of correction for SO2-based analyses. In: Report of sulfur isotope working group 8th advisory group meeting on future trends in stable isotope reference materials and laboratory quality assurance. Vienna, Austria: IAEA |
| [31] | Thode H G, Monster J, Dunford H B. 1961. Sulphur isotope geochemistry. Geochimica et Cosmochimica Acta, 25(3): 159-174 |
| [32] | Vanhaecke F, Dams R, Vandecasteele C. 1993.‘Zone model’as an explanation for signal behaviour and non-spectral interferences in inductively coupled plasma mass spectrometry. Journal of Analytical Atomic Spectrometry, 8(3): 433-438 |
| [33] | Weyer S, Schwieters J B. 2003. High precision Fe isotope measurements with high mass resolution MC-ICPMS. International Journal of Mass Spectrometry, 226(3): 355-368 |
| [34] | Xu J Q, Balik D, Agnes G R. 2001. Aerosol static electrification and its effects in inductively coupled plasma spectroscopy. Journal of Analytical Atomic Spectrometry, 16(7): 715-723 |
| [35] | Yang L. 2009. Accurate and precise determination of isotopic ratios by MC-ICP-MS: A review. Mass Spectrometry Reviews, 28(6): 990-1011 |
| [36] | 朱祥坤, 李志红, 赵新苗, 唐索寒, 何学贤, Belshaw N S. 2008. 铁同位素的MC-ICP-MS测定方法与地质标准物质的铁同位素组成. 岩石矿物学杂志, 27(4): 263-272 |