 2017, Vol. 44
2017, Vol. 44文章信息
- 影响癌症免疫检查点阻断药物治疗疗效和耐药的相关因素分析
- Factors Which Affect Efficacy and Resistance of Immune-checkpoint Blockade on Cancer
- 肿瘤防治研究, 2017, 44(8): 552-557
- Cancer Research on Prevention and Treatment, 2017, 44(8): 552-557
- http://www.zlfzyj.com/CN/10.3971/j.issn.1000-8578.2017.16.1488
- 收稿日期: 2016-12-02
- 修回日期: 2017-02-02
引用本文 |



2. 438000 黄冈,黄冈市中医医院肿瘤科
2. Department of Oncology, Huanggang Hospital of TCM, Huanggang 438000, China
肿瘤微环境(tumor microenvironment, TME)中免疫系统和癌症之间的相互作用,导致免疫耐受[1],包括调节性免疫细胞、免疫细胞因子和趋化因子,以及下调免疫功能的免疫检查点。在过去10年,癌症免疫治疗发展迅速,尤其是免疫检查点阻断药物(immune-checkpoint blocker, ICB),已经彻底改变了多种(以前认为预后不佳)恶性肿瘤的临床治疗,两个最突出的靶点是细胞毒T淋巴细胞相关蛋白4(CTLA-4)和PD-1/PD-L1。
迄今为止,六个ICB已被FDA批准:(1)Ipilimumab、CTLA-4阻断单抗,用于治疗不能手术切除或转移性黑色素瘤;(2)Pembrolizumab、PD-1阻断单抗,用于治疗不可切除的转移性黑色素瘤、PD-L1阳性的晚期转移性非小细胞肺癌(NSCLC)、复发难治性经典型霍奇金淋巴瘤、转移性尿路上皮癌和复发或转移性头颈部鳞癌;(3)Nivolumab、PD-1阻断单抗,用于治疗不能手术切除或转移性黑色素瘤、晚期转移性NSCLC(二线化疗)、晚期(转移性)肾细胞癌、复发难治性经典型霍奇金淋巴瘤、复发或转移性头颈部鳞癌和局部晚期或转移性尿路上皮癌;(4)Atezolizumab、PD-L1阻断单抗,批准用于治疗局部晚期或转移性尿路上皮癌;(5)Avelumab、PD-L1阻断单抗,用于治疗转移性Merkel细胞癌和晚期或转移性尿路上皮癌;(6)Durvalumab、PD-L1阻断单抗,用于治疗晚期或转移性尿路上皮癌。
以Nivolumab为例,不同肿瘤单药治疗反应率分别为黑色素瘤40%、非小细胞肺癌(NSCLC)20%、肾细胞癌25%、经典型霍奇金淋巴瘤66%和尿路上皮癌19.6%[2-5];提示大部分患者对ICB治疗无反应。因此,本文拟探讨以下两个问题:(1)ICB治疗存在明显异质性的原因、影响ICB疗效和耐药的因素,具体见表 1;(2)如何更有效并针对性的应用ICB。
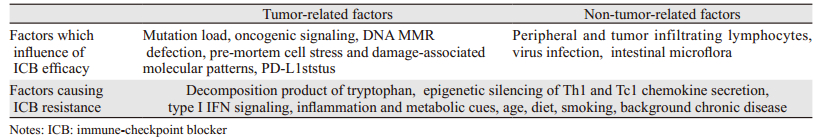 |
TME中T细胞完成主要的免疫监视,必须进行以下三个过程:(1)在淋巴结被适当的肿瘤抗原呈递细胞-树突细胞(DC)激活;(2)归巢到肿瘤,即从血管渗出,并穿透屏障如基质组织,到达恶性细胞周围;(3)识别并针对靶点发挥作用(通过CTL释放细胞溶解颗粒杀死恶性细胞)。进化的肿瘤通常积极阻止上述一个或多个T细胞免疫监视需要的过程,从而逃避免疫调节的肿瘤抑制[2]。
1.1 突变负荷基因的插入、删除和重排,除了能够影响肿瘤细胞生物学行为,还有编码新抗原的能力。高突变负荷的肿瘤可能具有更多的新抗原,因此免疫检查点阻断后,更容易激活肿瘤特异性T细胞[6]。平均突变负荷高的肿瘤(黑色素瘤、非小细胞肺癌、头颈部癌、膀胱癌和胃癌),对抗PD-1或抗PD-L1治疗反应率超过15%,而黑色素瘤高达30%~40%[7]。相反,平均突变负荷相对较低的癌症,如胰腺癌和前列腺癌,对抗PD-1治疗反应率低。而且,在有些类型的癌症中,不同病例的突变负荷差异可达百倍。
然而,目前没有明确的突变负荷界值可作为治疗参考,具有非常高突变负荷的患者可能没有反应,而突变负荷低的患者也可能有反应,关键在于新突变抗原肽是否具有肿瘤抗原。相对少数量的突变可以产生一个完美的抗原肽,而大量的突变却不一定会产生任何高品质的抗原肽;另一种情况是突变相关新抗原肽与T细胞耐受的自身抗原类似[8];最后,局部TME是影响免疫效应细胞功能的重要因素。
1.2 致癌信号癌细胞的自身致癌信号通路可能参与肿瘤微环境的共抑制分子表达,影响免疫治疗的效果。在黑色素瘤细胞中,致癌的WNT-B-catenin信号通路激活,抑制趋化因子CCL4,导致TME中缺乏T细胞和CD103+DC渗透,出现抗PD-L1和CTLA-4单抗治疗的耐药[9];在人神经胶质瘤中,高PD-L1表达和免疫抵抗相关[10];在霍奇金淋巴瘤中,染色体区域9p24.1的改变,通过Janus激酶(JAK)-信号转导和激活剂转录(STAT)信号通路,可诱导PD-L1的表达。此外,EGFR通路激活与免疫抑制之间存在相关[10-11],表现为PD-1、PD-L1/CTLA-4和炎性反应细胞因子上调,与低CTL和高T细胞耗竭相关,而阻断PD-1可恢复效应T细胞的功能,延长小鼠生存时间。
肿瘤细胞下调MHCⅠ类分子和TAP缺陷是另一种常见的肿瘤免疫逃逸机制[11]。与野生型比较,KRAS突变的肺腺癌亚群显示更高的PD-L1表达和炎性反应密度[12]。然而,在黑色素瘤,常见的致癌BRAF V600E突变似乎与PD-L1表达无关[13]。但是,相比单独的驱动基因突变,大量突变负荷可能对肿瘤免疫微环境具有更大的作用。
1.3 DNA错配修复(MMR)缺陷六个编码DNA错配修复复合体(MMR)的基因最初发现于Lynch综合征,后来发现除结直肠癌(colorectal cancer, CRC)之外,也存在于散发性癌症,包括胃癌、子宫内膜癌、壶腹、十二指肠、甚至前列腺癌中。MMR缺陷导致微卫星不稳定性(MSI),相比MMR完好的同种癌症,MMR缺陷患者的突变负荷升高10~100倍。事实上,MSI结肠癌有高度浸润的T细胞,尤其是CD8+T细胞。MSI结肠肿瘤中的CD4+肿瘤浸润淋巴细胞(tumor infiltrating lymphocyte, TIL)细胞向Th1型倾斜,可以产生大量干扰素γ。为了保持适应性抵抗,MSI肿瘤表达高水平的多个免疫调控分子,包括PD-1和PD-L1、CTLA4、淋巴细胞活化基因3(LAG3)。此外,还表达高水平IFN-γ诱导的免疫抑制代谢酶IDO1[14]。
在一项Pembrolizumab的三分组临床试验中,研究者发现约60%具有MSI的CRC和非结直肠癌患者有客观反应率,而微卫星稳定(microsatellite stability,MSS)患者没有反应[15]。DNA MMR基因型与抗PD-1治疗临床反应率之间的相关性,独立于肿瘤组织学类型,MSI很可能作为抗PD-1治疗疗效预测的生物标志物。
1.4 死亡前细胞应激和损伤相关分子模式内质网(ER)应激和自噬决定了肿瘤细胞的免疫原性死亡[16],影响损伤相关分子模式(DAMP)的表达或释放,募集特异的抗原呈递细胞(antigen presenting cell, APC)(如DC),识别和吞噬垂死的细胞成分,以便于处理和交叉呈递肿瘤抗原到同源T细胞。例如,TIL阴性的癌基因驱动的原发肺癌,对ICB和非免疫原性细胞毒(基于顺铂或紫杉烷的使用)治疗,以及二者联合治疗,均无反应。相反,ICB治疗前进行适当的化疗,能够形成更好的T细胞浸润的免疫微环境(即恢复Teff细胞/Treg细胞的比例),改善ICB疗效[17]。
最近一些研究发现,几个肿瘤相关的DAMP暴露——尤其是钙网蛋白膜表面暴露,TLR4配体HMGB1的释放和在肿瘤细胞胞质中LC3B荧光斑点的丰度增加(是自噬活性的替代标志)——与TIL浸润和治疗有效相关[18]。因此提示,肿瘤内细胞死亡的形式影响ICB疗效。
1.5 PD-L1的表达PD-L1通常由巨噬细胞的一个亚型表达,并可在活化的淋巴细胞(T、B细胞和NK细胞)、内皮细胞和其他非恶性细胞中被诱导,抑制免疫反应。然而,某些癌症中(如黑色素瘤、乳腺癌、头颈部鳞状细胞癌和肾癌),肿瘤细胞表面以及免疫细胞浸润细胞均表达PD-L1。结直肠癌和胃癌中,PD-L1几乎只表达于肿瘤浸润免疫细胞,很少表达于肿瘤细胞本身;而MCC,为中间类型。大样本的Nivolumab研究显示,PD-L1+肿瘤患者(至少5%的肿瘤细胞与免疫细胞表面表达PD-L1)治疗的反应是整体研究对象的两倍。随后在NSCLC、黑色素瘤、头颈部鳞状细胞癌、肾癌、膀胱癌中发现,PD-1通路阻断的总体反应率为29%,而PD-L1阳性的反应率为48%,PD-L1阴性为15%。虽然PD-L1表达和抗PD-1治疗的长期结果如无进展生存和总体生存的关系尚未确立,但一些研究认为,PD-L1表达的肿瘤患者生存期延长[19]。
2015年10月,PD-L1 IHC 22C3 pharm检测作为Pembrolizumab治疗晚期NSCLC的伴随诊断获得FDA批准。源于一项Ⅱ期临床试验发现,在NSCLC中,与低PD-L1表达水平相比,表达率≥50%的PD-L1阳性患者(约占20%)对抗PD-1治疗有更高的反应率,且无进展生存期和总生存期延长。然而,抗PD-1药物Nivolumab的两项Ⅲ期临床试验中,使用PD-L1 IHC 28-8 pharmDx检测,发现治疗前肿瘤细胞表达PD-L1的非鳞癌患者,总生存期延长;但鳞癌未显示此结果。因此,2015年10月,FDA批准PD-L1 IHC28-8pharmDx检测作为Nivolumab治疗肺癌的一种补充而非必要诊断测试,随后在2016年1月批准作为Nivolumab治疗黑色素瘤的补充检测,以PD-L1 > 1%的临界值界定为阳性结果[20]。然而,虽然PD-L1阴性肿瘤患者缓解率较低,不能作为治疗的绝对选择标准,但是,在可选择多种治疗方案的患者中,PD-L1表达可能用于优选治疗顺序,即PD-L1阳性肿瘤建议一线抗PD-1/PD-L1治疗,而PD-L1阴性的肿瘤患者则作为二线或多线后治疗。但是,多项研究试验结果不尽相同,提示采用IHC 22C3和IHC 28-8 PD-L1检测,还需要更多的临床数据支持。
PD-L1蛋白表达的精确测量和评分也存在各种技术和生物性障碍。例如商品化自动免疫组织化学的检测方法不统一,检测PD-L1所使用的单抗不同;PD-L1表达具有时间异质性,比如抗PD-1治疗开始前数天、数月或数年的肿瘤标本,其PD-L1的表达也有变化。此外,IHC技术有许多变量,包括抗原修复条件、温度、抗体浓度和孵育时间、检测系统。组织类型和固定时间,也是重要的变量。这些都将影响免疫组织化学测定结果。此外,符合临床试验的患者先前通常接受多种治疗,也可能会改变肿瘤的免疫微环境。
2 影响免疫检查点阻断的非肿瘤相关因素 2.1 外周和肿瘤内淋巴细胞与PD-1调节抗原表达效应细胞不同,CTLA-4影响广泛,通过抑制CD4+T效应(Teff)细胞和增强Treg细胞活性参与免疫反应启动。因此,抗CTLA-4治疗(Ipilimumab, Tremelimumab)的许多生物标志的研究都集中在治疗前后淋巴细胞的多样性、表型和功能。
多个研究指出,外周血淋巴细胞绝对数上升与较高的Ipilimumab治疗反应率有关。此外,具有抗NY-ESO-1癌-睾丸抗原的CD4+和CD8+T淋巴细胞特异性的黑色素瘤患者,也表现出明显的肿瘤缩小或稳定[21]。相反,外周血中高水平可溶性CD25(也称为IL2Rα),与抗CTLA-4治疗耐药有关[22]。治疗前TME的PD-L1表达,一般与抗CTLA-4治疗临床反应无关。多种类型的肿瘤在CTLA-4阻断后,PBL和TIL上诱导性T细胞共刺激分子(ICOS)表达增加,且肿瘤组织中Treg细胞比例增加。
在人类CRC标本中,发现高密度的CD3+CD8+ CD45RO+颗粒酶+T细胞与低肿瘤复发和高总生存率有关。在某些情况下,三级淋巴结构(Tertiary lymphoid structure),可以促进T细胞的募集,并协调局部自适应抗肿瘤免疫反应,进而改善患者预后。具体地说,在黑色素瘤的抗PD-1治疗中,侵袭性肿瘤边缘的CD8+T细胞密度与抗PD-1疗效相关[23]。然而,目前没有Ipilimumab治疗的预测性生物标志应用于临床,这可能与TME免疫抑制有关。耗竭Treg细胞,可以与ICB治疗联合使用,重新恢复现有T细胞杀灭肿瘤的能力。
2.2 病毒相关的因素在癌症基因组中,致癌病毒可能赋予新抗原性,因此可作为分子标志,预测肿瘤对检查点阻断的反应。某些肿瘤的发生几乎均与病毒相关,例如鼻咽癌(EBV)、宫颈癌和肛门癌(HPV)、成人T细胞白血病(HTLV-1)和卡波济肉瘤(KSHV)。而有些肿瘤,如胃癌(EBV)、霍奇金淋巴瘤(EBV)、MCC(MCPyV)和头颈部鳞状细胞癌(HPV),只有某些亚型与病毒相关。此外,大多数肝细胞癌是由乙型肝炎病毒(乙肝病毒)或丙型肝炎病毒的慢性感染造成的。重要的是,与肿瘤基因组的点突变或重排能够产生单个或数量有限的被T细胞识别的抗原肽不同,源于病毒的基因,编码的整个蛋白产物都是非己的,从而存在许多潜在的T细胞表位。
病毒相关癌症对抗PD-1治疗有高反应率。初步数据显示,晚期MCC约80%由病毒导致,对抗PD-1治疗(Pembrolizumab)的反应率超过50%。也有报道HBV和HCV相关的肝细胞癌对抗PD-1治疗有明显反应[24]。
2.3 肠道微生物因素最近的研究表明,肠道菌群与宿主具有互惠互利的共生关系。虽然每一个体的微生物群组成总体是稳定的,但个体间存在巨大的异质性(即使同卵双胞胎之间),因此,肠道菌群代表人类一个显著的遗传和代谢多样性。
宿主和肠道细菌之间的互惠共生失衡,可以导致多种自身炎性反应和自身免疫性疾病,如肥胖、糖尿病、炎性肠疾病和非酒精性脂肪肝病,以及多种癌症(结直肠癌或肠外癌症,如乳腺癌和肝癌)。
肠道菌群也影响肿瘤免疫治疗疗效。最近的证据表明,CTLA-4阻断剂,通过诱导明显的杆菌属细菌在黏液层内侧积累,并与黏膜DC接触,激活IL-12依赖的Th1细胞免疫反应,有利于宿主抗肿瘤反应。Ipilimumab治疗的转移性黑素瘤患者,存在三种不同的微生物型(肠型,enterotypes),由杆菌和普雷沃属决定(Alloprevotella和普氏菌属于A型、类杆菌属B和C型)。粪便微生物组合C,含有丰富的免疫原性杆菌属(主要是移植有助于脆弱拟杆菌的小生境),可以恢复对CTLA-4单抗的疗效,而组合物B,含丰富种杆菌,与全面的单抗耐药有关[25]。同样,研究已证明,双歧杆菌导致TME内TIL的富集增强PD-L1单克隆抗体的抗肿瘤效果。由此可见,肠道微生物在ICB最佳治疗中的重要性,但不同的细菌导致不同的效果。
3 影响免疫检查点阻断药物耐药的相关因素 3.1 色氨酸的分解产物TME内色氨酸的分解代谢日益被认为参与导致抑制抗肿瘤免疫反应。色氨酸通过限速酶IDO(表达于骨髓细胞和癌症细胞)分解代谢,产生免疫抑制的代谢产物,如犬尿氨酸,抑制T细胞克隆增殖,诱导T细胞无反应和细胞凋亡[26]。因此,IDO抑制剂和ICB治疗联合,已在实验中证明能增加TME的TIL及其功能,从而导致IDO表达和不表达的弱免疫原性肿瘤的消退。
3.2 表观遗传沉默趋化因子分泌TME的基质细胞可以阻止T细胞与癌细胞直接相互作用。由于TME调节肿瘤内T细胞的局部增殖能力,控制T细胞活性,调节与癌细胞相关的T细胞空间分布,或通过TME控制T细胞从循环系统向肿瘤的迁移[1]。如编码趋化因子CXCL9和CXCL10——直接指引T细胞向肿瘤迁移的基因出现后天沉默。事实上,使用表观遗传调节剂治疗,可以解除对Th1细胞分泌趋化因子的抑制,并且增加TIL,因此能够提高PD-L1检查点阻断治疗的效果。
3.3 Ⅰ型干扰素信号转导TME明显缺乏可分泌Ⅰ型干扰素的DC,自然导致ICB治疗时,抗肿瘤T细胞启动不足和T细胞重新激活的数目有限。而敏感通路的充分激活(如TLR3-IFNAR刺激,蒽环类化疗诱导肿瘤细胞免疫原性死亡[ICD])能诱导Ⅰ型IFN,然后反过来又刺激趋化因子CXCL10的分泌,募集TIL进入瘤床。同样,体内研究已经证实,BATF3型DC通过激活干扰素活化基因(STING)通路,生产Ⅰ型IFN,在诱导最佳的T细胞募集至肿瘤和自发抗肿瘤T细胞反应中,发挥必需的重要作用[27]。在免疫细胞贫乏的黑色素瘤,体内肿瘤周围注入免疫调节RNA,通过各种免疫细胞分泌的Ⅰ型IFN驱动,能够抑制肿瘤生长。随后进行PD-1/PD-L1靶向阻断治疗,Ⅰ型IFN激活会增强抗肿瘤活性。联合CTLA-4阻断与干扰素α-2b的初步临床试验已经表现出一些阳性结果可能。
3.4 炎性反应与代谢因素在TME内新陈代谢和炎性反应过程可以参与抑制所期望的ICB疗效。最近发现,肿瘤细胞环氧合酶(COX)通过其产生的前列腺素E2发挥免疫抑制功能,该功能导致的抑制免疫和促进炎性反应环境有利于肿瘤生长[28]。同样,在TME中肿瘤细胞的葡萄糖消耗可以导致对T细胞产生代谢限制、抑制mTOR活性、糖酵解能力,以及T细胞内IFN-γ的生产,引起肿瘤进展。因此预测,对糖酵解率较高的肿瘤患者,ICB可能是最有效的治疗。有研究显示,胆固醇代谢的调控也可以被用来加强CD8+T细胞的抗肿瘤反应。抑制胆固醇酯化反应可以促进CD8+T细胞(但不是CD4+T细胞)有效控制小鼠黑素瘤的生长和转移。
3.5 年龄众所周知,衰老机体的免疫功能下降。癌症免疫治疗必须调动一个衰老的免疫系统的功能。年龄相关的免疫系统改变包括:(1)APC及其功能的下降;(2)淋巴细胞数目减少,因此可能降低克隆异构以维持免疫监视;(3)出现具有终末分化标志物的T细胞,难以激活;(4)慢性炎性反应信号激活,包括促炎细胞因子的循环水平,如IL-1、IL-6和肿瘤坏死因子(TNF)增加;(5)抑制性免疫细胞的数目激增,包括肿瘤内MDSC和Treg细胞。总之,明显的免疫衰老有可能形成一个强大的ICB治疗屏障,所以最好先进行老年患者免疫功能的评估,再选择治疗方法。
3.6 饮食和吸烟通常建议接受免疫治疗的患者维持健康的饮食,摄取足够的维生素和矿物质以增强免疫系统的功能。对结直肠癌患者的研究中发现,提供脂溶性抗氧化剂可促进CD4+T细胞数量,以及Th1细胞的功能。在TME中,免疫治疗可能受到宿主能量代谢相关因素的影响,但相关研究不多。NSCLC中,吸烟者有较高的PD-1阻断治疗的反应率,可能是因为吸烟相关肺癌的突变负荷要远高于非吸烟者。
3.7 慢性感染除了TME相关的抑制,慢性病毒感染导致的免疫信号紊乱也可以影响T细胞浸润。如慢性丙型肝炎病毒(HCV)感染与CXCL10的N末端截短形式强烈相关,其通过拮抗CXCR3(与CXCL10结合)调节的信号,抑制效应T细胞和NK细胞趋化到病毒感染的细胞和肿瘤[29]。二肽基肽酶4(DPP4)可产生截断形式的拮抗性CXCL10两者均与HCV感染者明显的肝脏疾病和治疗失败相关。同时,DPP4能够抑制T细胞向肿瘤迁移,而抑制DPP4可改善辅助的免疫治疗、过继T细胞回输和检查点阻断的效果。此外,其他慢性感染导致的类似免疫信号也有可能会影响肿瘤免疫治疗。
4 联合治疗基于阻断免疫检查点的药物,联合其他免疫检查点阻断剂或抗癌药物的治疗方案的开发是目前的热门。事实上,在接下来的5~10年,超过50个免疫ICB的联合方法将被尝试应用,如细胞因子或激动剂(如GM-CSF、干扰素α-2b和TLR激动剂)、IDO抑制剂、基于肽的疫苗、靶向疗法、溶瘤病毒治疗、化学治疗、放射治疗或其他ICB,目前均在临床试验阶段。许多联合策略都可能产生成功,然而,同样推测毒性反应可能明显增加。
依据临床前证据,抗PD-1/PD-L1和(或)抗CTLA-4的有效联合治疗可能包括放射治疗、特定的化疗、激酶抑制剂或表观遗传的药物,后者本身已被证明具有免疫原性。此外,由于潜在的靶点序列和作用机制不同,可以联合两种或以上不同的免疫检查点阻断药物。在PD-L1的黑色素瘤,与Nivolumab单药治疗相比,Ipilimumab和Nivolumab联合治疗的无进展生存期更长;然而,在PD-L1阳性的肿瘤患者中疗效似乎相当。提示PD-L1检测可能对Nivolumab治疗选择有用,从而避免Nivolumab联合Ipilimumab联合治疗出现相当高严重毒性的发生率。2015年10月,FDA批准Ipilimumab加Nivolumab治疗晚期BRAF野生型黑色素瘤,这是免疫治疗药物联合方案被首次批准,具有里程碑意义。
个体间肠道微生物的差异是免疫治疗疗效和毒性异质性的一个原因,提示有可能通过辅助癌微生物(oncomicrobiotics),即提供共生菌群和(或)抗生素选择性除掉免疫抑制性微生物的活免疫原性共生细菌,提供含有与癌症抗原分子高度相似的细菌抗原成分,改善ICB治疗中T细胞的激活指数[30-31]。近来,MetaHIT和人类微生物群项目的宏基因组学和宏翻译组学报道了人类肠道微生物参考基因全目录,同时联合环境组学和MALDI-TOF质谱法识别新共生菌。肿瘤学家有朝一日可以利用这些资料库,进一步探索癌症相关的微生物。
5 结论及展望随着对癌症TME和全身免疫调节的理解加深,影响ICB治疗效果的各种因素都得到了人们的研究,很可能会发现更多的生物标志物。肿瘤细胞和T细胞的代谢竞争葡萄糖,提示代谢生物标志物可能是抗肿瘤反应的关键因素。另一个潜在的生物标志物是肠道微生物,可能会提高ICB治疗的反应;然而,是哪种益生菌和特定细菌株构成状态,仍有待进一步研究。最后,与直接靶向抑制激酶活性的驱动突变定义的静态生物标志不同,不断变化的免疫系统为生物标志物的发展提出了独特的挑战。
| [1] | Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment[J]. Science, 2015, 348(6230): 74–80. DOI:10.1126/science.aaa6204 |
| [2] | Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation[J]. N Engl J Med, 2015, 372(4): 320–30. DOI:10.1056/NEJMoa1412082 |
| [3] | Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer[J]. N Engl J Med, 2015, 373(2): 123–35. DOI:10.1056/NEJMoa1504627 |
| [4] | Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma[J]. N Engl J Med, 2015, 373(19): 1803–13. DOI:10.1056/NEJMoa1510665 |
| [5] | Younes A, Santoro A, Shipp M, et al. Nivolumab for classical Hodgkin's lymphoma after failure of both autologous stem-cell transplantation and brentuximab vedotin: a multicentre, multicohort, single-arm phase 2 trial[J]. Lancet Oncol, 2016, 17(9): 1283–94. DOI:10.1016/S1470-2045(16)30167-X |
| [6] | Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity incancer and the search for new cancer-associatedgenes[J]. Nature, 2013, 499(457): 214–8. |
| [7] | Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer[J]. N Engl J Med, 2012, 366(26): 2455–65. DOI:10.1056/NEJMoa1200694 |
| [8] | Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma[J]. N Engl J Med, 2014, 371(23): 2189–99. DOI:10.1056/NEJMoa1406498 |
| [9] | Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic b-cateninsignalling prevents anti-tumour immunity[J]. Nature, 2015, 523(559): 231–5. |
| [10] | Akbay EA, Koyama S, Carretero J, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors[J]. Cancer Discov, 2013, 3(12): 1355–63. DOI:10.1158/2159-8290.CD-13-0310 |
| [11] | Haworth KB, Leddon JL, Chen CY, et al. Going back to class Ⅰ: MHC and immunotherapies for childhood cancer[J]. Pediatr. Blood Cancer, 2015, 62(4): 571–6. DOI:10.1002/pbc.25359 |
| [12] | Skoulidis F, Byers LA, Diao L, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities[J]. Cancer Discov, 2015, 5(8): 860–77. DOI:10.1158/2159-8290.CD-14-1236 |
| [13] | Rodić N, Anders RA, Eshleman JR, et al. PD-L1 expression in melanocytic lesions does not correlate with the BRAF V600E mutation[J]. Cancer Immunol Res, 2015, 3(2): 110–5. DOI:10.1158/2326-6066.CIR-14-0145 |
| [14] | Llosa NJ, Cruise M, Tam A, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints[J]. Cancer Discov, 2015, 5(1): 43–51. DOI:10.1158/2159-8290.CD-14-0863 |
| [15] | Le DT, Uram JN, Wang H, et al. PD-1 Blockade in tumors with mismatch-repair deficiency[J]. N Engl J Med, 2015, 372(26): 2509–20. DOI:10.1056/NEJMoa1500596 |
| [16] | Galluzzi L, Buqué A, Kepp O, et al. Immunological effects of conventional chemotherapy and targeted anticancer agents[J]. Cancer Cell, 2015, 28(6): 690–714. DOI:10.1016/j.ccell.2015.10.012 |
| [17] | Pfirschke C, Engblom C, Rickelt S, et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy[J]. Immunity, 2016, 44(2): 343–54. DOI:10.1016/j.immuni.2015.11.024 |
| [18] | Fucikova J, Becht E, Iribarren K, et al. Calreticulin Expression in Human Non-Small Cell Lung Cancers Correlates with Increased Accumulation of Antitumor Immune Cells and Favorable Prognosis[J]. Cancer Res, 2016, 76(7): 1746–56. DOI:10.1158/0008-5472.CAN-15-1142 |
| [19] | Sunshine J, Taube JM. PD-1/PD-L1 inhibitors[J]. Curr Opin Pharmacol, 2015, 23: 32–8. DOI:10.1016/j.coph.2015.05.011 |
| [20] | Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer[J]. N Engl J Med, 2015, 373(17): 1627–39. DOI:10.1056/NEJMoa1507643 |
| [21] | Yuan J, Adamow M, Ginsberg BA, et al. Integrated NY-ESO-1 antibody and CD8+ T-cell responses correlate with clinical benefit inadvanced melanoma patients treated with ipilimumab[J]. Proc Natl Acad Sci U S A, 2011, 108(40): 16723–8. DOI:10.1073/pnas.1110814108 |
| [22] | Hannani D, Vétizou M, Enot D, et al. Anticancer immunotherapy by CTLA-4 blockade: obligatory contribution of IL-2 receptors and negative prognostic impact of soluble CD25[J]. Cell Res, 2015, 25(2): 208–24. DOI:10.1038/cr.2015.3 |
| [23] | Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome[J]. Science, 2006, 313(5795): 1960–4. DOI:10.1126/science.1129139 |
| [24] | Melero I, Crocenzi TS, Weling TH, et al. PhaseⅠ/Ⅱsafety and antitumor activity of nivolumab in patients with advanced hepatocellular carcinoma (HCC): CA209-040[J]. J Clin Oncol, 2015, 33(15 suppl): LBA101. |
| [25] | Sivan A, Corrales L, Hubert N, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy[J]. Science, 2015, 350(6264): 1084–9. DOI:10.1126/science.aac4255 |
| [26] | Platten M, Wick W, Van den Eynde B J. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion[J]. Cancer Res, 2012, 72(21): 5435–40. DOI:10.1158/0008-5472.CAN-12-0569 |
| [27] | Corrales L, Gajewski TF. Molecular pathways: targeting the stimulator of interferon genes (sting) in the immunotherapy of cancer[J]. Clin Cancer Res, 2015, 21(21): 4774–9. DOI:10.1158/1078-0432.CCR-15-1362 |
| [28] | Zelenay S, van der Veen AG, Böttcher JP, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity[J]. Cell, 2015, 162(6): 1257–70. DOI:10.1016/j.cell.2015.08.015 |
| [29] | Casrouge A, Decalf J, Ahloulay M, et al. Evidence for an antagonist form of the chemokine CXCL10 in patients chronically infected with HCV[J]. J Clin Invest, 2011, 121(1): 308–17. DOI:10.1172/JCI40594 |
| [30] | Sivan A, Corrales L, Hubert N, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy[J]. Science, 2015, 350(6264): 1084–9. DOI:10.1126/science.aac4255 |
| [31] | Viaud S, Saccheri F, Mignot G, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide[J]. Science, 2013, 342(6161): 971–6. DOI:10.1126/science.1240537 |