



2. 河南省中西医结合医院中药研究所,河南 郑州 450052
2. Henan Integrative Medicine Hospital, Institute of Chinese Medicine, Zhengzhou 450052, China
UDP-葡萄糖醛酸转移酶(UDP-glucuronosyl transferases, UGTs)作为一类重要的肝脏药物代谢酶[1],在药物代谢和清除过程中扮演着关键角色。精确评估药物对UGTs的抑制潜力对于药物研发和临床合理用药指导极为重要。根据国家药品监督管理局、美国食品药品监督管理局和人用药品技术要求国际协调理事会颁布的药物相互作用研究相关指导原则,应在首次人体试验之前,开展体外代谢试验评估Ⅰ相代谢酶肝细胞色素(cytochrome,CYP)P450酶和Ⅱ相代谢酶UGTs酶与在研药物之间相互作用的可能性,为临床药动学研究设计提供参考。虽然国内已有报道药物对人肝CYP450酶诱导和抑制作用的体外评价体系[2],但尚未有UGTs酶活性与体外抑制评价体系的相关报道。因此建立体外评价体系以评估药物对UGTs的抑制潜力,并提供有价值的信息以预测体内药物间的相互作用至关重要。虽然国外已开发出多种方法来评价UGTs的抑制作用,包括代谢产物生成法、基于荧光的测定法和放射色谱法等。然而,上述方法存在一些缺陷,如测量准确性和稳定性的不足,以及缺乏经过验证的分析方法来量化底物的代谢产物[3]。因此,建立一个可靠且准确的体外评价体系,以评估肝脏UGTs酶活性及药物对人肝UGTs的抑制作用,仍然具有挑战。液相色谱-串联质谱法(liquid chromatography-tandem mass spectrometry, LC-MS/MS)因其出色的灵敏度、高选择性和广泛的动态测量范围,在复杂体系中测定小分子药物及其代谢产物定量分析方面,展现出了无可比拟的优势。此外,该方法避免了传统酶活性检测方法(如放射同位素标记或色谱法)的一些不足,如需要专门废物处理设备来处理放射性废物以及交叉反应问题。
本研究旨在建立一个可靠准确的体外评价体系,利用经过方法学确证的强特异性和高灵敏度的LC-MS/MS方法,评估肝脏UGTs酶活性,并通过已知的UGTs抑制剂的抑制效果验证评价体系的可行性。成功建立此评价体系将为药物开发和合理临床用药提供重要指导,确保药物的安全有效使用,同时将药物间的相互作用风险降到最低。
1 材料与方法 1.1 仪器和试剂ExionLCTM液相色谱仪、SCIEX Triple QuadTM 4500三重四极杆质谱仪、Analyst Software 1.6.3数据采集软件、MultiQuant 3.0.2数据处理软件(美国Applied Biosystems公司);XPE205型电子分析天平(瑞士METTLER TOLEDO公司);Bio Tool D3415R型低温高速离心机(瑞士Bio Tool公司);微量移液器(德国Eppendorf公司);VORTEX QL901涡旋振荡器(中国Kylin-Bell Lab Instruments公司)。
对照品7-乙基-10-羟基喜树碱(7-Ethyl-10-hydroxy-camptothecin,SN-38,纯度99%),鹅去氧胆酸(chenodeoxycholic acid,纯度98%),三氟拉嗪(trifluoperazine,纯度98%),霉酚酸(mycophenolic acid, 纯度>95%),3'-叠氮化物-3'脱氧胸苷(3'-azide-3' deoxythymidine,zidovudine, 纯度98%),丙甲菌素(纯度>98%)和尿苷5'-二磷酸葡萄糖醛酸三钠盐(uridine 5'-diphosphoglucuronic acid trisodium salt,UDPGA,纯度>95%)购自中国百灵威科技有限公司,N-乙酰血清素(N-Acetylserotonin,纯度98%)购自梯希爱(上海)化成工业发展有限公司,7-乙基-10-羟基喜树碱葡糖苷酸(纯度97%),鹅去氧胆酸24-酰基-β-D-葡糖苷酸(纯度96%),N-乙酰血清素β-D-葡糖苷酸(纯度97%),霉酚酸β-D-葡糖苷酸(纯度96%),3′-叠氮化物-3′-脱氧胸苷β-D-葡糖苷酸钠盐(纯度97%),呋喃茶碱(纯度98%),购自加拿大Toronto Research Chemicals Inc公司;三氟拉嗪N-葡糖苷酸(纯度>96%)购自TLC Pharmachem。地西泮注射液购自天津金耀药业股份有限公司。合并的肝微粒体购自美国Corning Life Sciences。
试验用水为MilliQ纯水;甲醇、乙腈(色谱纯)购自美国Fisher Chemical公司;其他试剂为分析纯。
1.2 色谱及质谱条件色谱柱:菲罗门Kinetex C18色谱柱(2.1 × 100 mm, 2.6 μm);流动相:甲醇-水(含0.1%甲酸)梯度洗脱,洗脱比例如下:0~1 min,20%甲醇;1~5 min,20~80%甲醇;5~7 min,80%甲醇;7~8 min,80~20%甲醇;8~9.5 min,20%甲醇,流速:300 μL·min-1;柱温:40 ℃。分析时间:9.5 min。
离子源:ESI+/ESI-;检测方法:正/负离子模式;扫描方法:多反应监测(MRM);去溶剂气:N2;离子源温度:550 ℃;离子喷雾电压:正离子模式:5 000 V,负离子模式:-4 500 V;窗帘气,碰撞气、离子源气1和离子源气2分别设置为35、7、40和65 psi。
1.3 人肝微粒体UGTs酶活性的评价肝微粒体孵育体系包含100 mmol·L-1 Tris-HCL缓冲液(pH=7.40)、5 mmol·L-1氯化镁、50 mg·L-1阿拉霉素、不同浓度的肝微粒体蛋白,1 mmol·L-1 UDPGA,不同浓度的探针底物。反应总体积为100 μL。将阿拉霉素和肝微粒体蛋白加入Tris-HCl缓冲液,置于冰上放置15 min,以利于阿拉霉素在微粒体表面形成模孔,暴露出UGT酶,15 min后,加入剩余组分。在37 ℃预孵育5 min后,加入UDPGA启动反应。然后在37 ℃进一步孵育一定时间后(详见Tab 1),加入含有内标(地西泮和呋喃茶碱)的100 μL乙腈终止反应。涡旋混合2 min后,4 ℃的14 000 r·min-1离心10 min。取上清100 μL进样,进样体积2 μL,测定温孵体系中生成的各底物相应的代谢产物浓度,按下式计算各代谢产物的生成速率: 代谢产物的生成速率=代谢产物的浓度/(微粒体蛋白浓度×温孵反应时间)。使用GraphPad Prism 6软件按照非线性回归计算各底物的米氏常数Km和Vmax。
| Enzyme | Substrate | Incubation condition | Enzyme activity Substrate concentration /μmol·L-1 | |
| Protein concentration /g·L-1 | Time /min | |||
| UGT1A1 | SN-38 | 0.1 | 60 | 0.1, 0.2, 0.5, 1, 2, 5, 10, 50, 100 |
| UGT1A3 | Chenodeoxycholic acid | 0.1 | 30 | 5, 10, 50, 100, 500, 1000, 1200 |
| UGT1A4 | Trifluoperazine | 0.05 | 10 | 1, 2, 5, 10, 20, 50, 100, 500 |
| UGT1A6 | N-acetylserotion | 0.025 | 6 | 10, 20, 50, 100, 200, 500, 1000, 1500 |
| UGT1A9 | Mycophenolic acid | 0.1 | 30 | 1, 2, 5, 10, 20, 50, 100, 500 |
| UGT2B7 | Zidovudine | 0.075 | 15 | 10, 20, 50, 100, 200, 500, 1000, 2000 |
将不同浓度的已知抑制剂(1.00 μL抑制剂溶液阳性对照)分别与底物探针共同温孵,底物和阳性对照抑制剂的浓度见Tab 2,对照组(不含抑制剂)加入等体积的缓冲液,余下按“人肝微粒体UGTs酶活性的评价”项下操作。按下式计算剩余率:剩余率=生成速率(含抑制剂)/ 生成速率(不含抑制剂)×100%。
| Enzyme | Substrate | Substrate concentration /μmol·L-1 | Inhibitor | Inhibitor concentration/ μmol·L-1 |
| UGT1A1 | SN-38 | 1 | Atazanavir | 0, 0.1, 0.2, 0.5, 1, 2 |
| UGT1A3 | Chenodeoxycholic acid | 20 | Celastrol | 0, 1, 5, 10, 20, 50, 100 |
| UGT1A4 | Trifluoperazine | 50 | Hecogenin | 0, 0.1, 0.2, 0.5, 1, 2 |
| UGT1A6 | N-Acetylserotion | 500 | Troglitazone | 0, 1, 5, 10, 20, 50, 100 |
| UGT1A9 | Mycophenolic acid | 500 | Niflumic acid | 0, 0.1, 0.2, 0.5, 1, 2 |
| UGT2B7 | Zidovudine | 500 | Mefenamic acid | 0, 1, 5, 10, 20, 50, 100 |
使用GraphPad Prism 6计算主要的酶促动力学参数。根据Michaelis-Menten公式(公式1)进行计算。公式中,Vmax表示最大反应速率;Km表示反应速率为Vmax一半时的底物浓度, S为底物浓度。
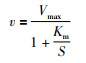
|
(1) |
以剩余率为纵坐标, 抑制剂浓度的对数值为横坐标, 绘制抑制曲线, 使用GraphPad Prism 5.01软件按照非线性回归, 计算各抑制剂的半数抑制浓度(50% inhibition concentration, IC50)。
2 结果 2.1 分析方法学验证 2.1.1 各待测物离子选择通道及对碰撞诱导解离电压和去簇电压如Tab 3所示。
| Enzyme | Analyte | MRM transition | DP/V | EP/V | CE/V | CXP/V |
| UGT1A1 | SN-38-glucuronide | 567.3 → 346.9 | -102.06 | -6.58 | -45.13 | -25.02 |
| UGT1A3 | Chenodeoxycholic acid-glucuronide | 567.4 → 391.3 | -45.03 | -4.10 | -47.03 | -25.31 |
| UGT1A4 | Trifluoperazine-glucuronide | 584.2 → 408.0 | 103.9 | 4.99 | 34.48 | 17.19 |
| UGT1A6 | N-acetylserotion-glucuronide | 395.1 → 219.0 | 59.83 | 7.86 | 17.69 | 8.88 |
| UGT1A9 | Mycophenolic acid-glucuronide | 495.2 → 319.0 | -35.96 | -11.18 | -26.54 | -22.07 |
| UGT2B7 | Zidovudine-glucuronide | 442.3 → 124.8 | -63.11 | -6.94 | -29.36 | -10.94 |
| IS | Furafylline | 258.9 → 177.8 | -76.38 | -4.85 | -24.19 | -13.28 |
| IS | Diazepam | 285.3 → 154.1 | 118.04 | 11.00 | 32.00 | 10.00 |
| DP: Declustering potential; EP: Entrance potential; CE: Collision energy; CXP: Collision cell exit potential; IS: Internal standard | ||||||
在本实验所采用的色谱条件下, 7-乙基-10-羟基喜树碱葡糖苷酸,鹅去氧胆酸24-酰基-β-D-葡糖苷酸,三氟拉嗪N-葡萄糖醛酸苷酸,N-乙酰血清素β-D-葡糖苷酸,霉酚酸β-D-葡糖苷酸,3′-叠氮化物-3′-脱氧胸苷β-D-葡糖苷酸,和内标(地西泮和呋喃茶碱)的保留时间见Fig 1。各待测组分及内标的峰形良好,无内源性干扰,方法具有较高的特异性。

|
| Fig 1 Chromatograms of blank HLM incubation system (a), blank HLM incubation system spiked with reference standards of SN-38-glucuronide (5 μg·L-1), chenodeoxycholic acid glucuronide (25 μg·L-1), trifluoperazine-glucuronide (2.5 μg·L-1), N-acetyl serotion-glucuronide (25 μg·L-1), mycophenolic acid-glucuronide (25 μg·L-1), zidovudine-glucuronide (25 μg·L-1), internal standard (furafylline), and internal standard (diazepam) (b), and extracted samples of SN-38 (1 μmol·L-1), chenodeoxycholic acid (5 μmol·L-1), trifluoperazine (1 μmol·L-1), N-acetylserotion (10 μmol·L-1), mycophenolic acid (1 μmol·L-1), zidovudine (10 μmol·L-1), internal standard (furafylline), and internal standard (diazepam) after enzymatic incubation (c). |
分别制备含各UGTs亚型底物代谢产物的标准曲线,进行LC-MS/MS分析,记录色谱图。分别计算样本峰面积As和内标峰面积Ai的比f(f=As/Ai),以峰面积比f对浓度作权重回归计算。各UGTs亚型底物代谢产物的典型回归方程见Tab 4。
| Analyte | SC range/μg·L-1 | QC level/μg·L-1 | Typical standard curve |
| SN-38-glucuronide | 1-100 | 2, 10, 80 | Y=0.001 33X-3.37660e-4, R=0.998 6 |
| Chenodeoxycholic acid glucuronide | 25-5 000 | 50, 100, 4000 | Y=8.452 47e-4X-0.00207, R=0.995 9 |
| Trifluoperazine-glucuronide | 2.5-500 | 5, 10, 400 | Y=0.016 02X+0.01140, R=0.996 7 |
| N-acetylserotion-glucuronide | 25-5 000 | 50, 100, 4000 | Y=0.001 76X+0.00217, R=0.997 5 |
| Mycophenolic acid-glucuronide | 25-5 000 | 50, 100, 4000 | Y=0.003 03X-1.97079e-4, R=0.997 3 |
| Zidovudine-glucuronide | 25-5 000 | 50, 100, 4000 | Y=1.537 35e-4X-1.74484e-, R=0.998 0 |
| SC: Standard curve; QC: Quality control | |||
分别制备含最低定量下限、低、中、高4种浓度各UGTs亚型底物代谢产物的标准含药样本,共考察3批。进行LC-MS/MS分析,计算实测浓度、准确度和批内批间精密度。批内和批间精密度的相对平均偏差(relative standard deviation,RSD)小于15%, 准确度的平均值在87.4%~112.1%内。详见Tab 5。
| Analyte | Concentration levels (x±s, μg·L-1) | RSD/% | RE/% Accuracy | |||
| Added | Measured | Intra-day | Inter-day | |||
| SN-38-glucuronide | 2 | 1.99 | 4.89 | 8.41 | 99.6 | |
| 10 | 8.74 | 2.80 | 6.96 | 87.4 | ||
| 80 | 90.38 | 4.86 | 8.97 | 113.0 | ||
| Chenodeoxycholic acid glucuronide | 50 | 50.87 | 5.30 | 5.76 | 101.7 | |
| 100 | 90.23 | 3.80 | 7.57 | 90.2 | ||
| 2 000 | 2 078.44 | 4.32 | 10.82 | 103.9 | ||
| Trifluoperazine-glucuronide | 5 | 5.29 | 1.91 | 7.17 | 105.8 | |
| 10 | 9.22 | 1.97 | 4.19 | 92.2 | ||
| 400 | 428.17 | 3.07 | 5.19 | 107.0 | ||
| N-acetylserotion-glucuronide | 50 | 50.23 | 5.03 | 6.05 | 100.4 | |
| 100 | 103.17 | 2.83 | 10.21 | 103.2 | ||
| 4 000 | 3768.66 | 4.26 | 5.40 | 94.2 | ||
| Mycophenolic acid-glucuronide | 50 | 48.59 | 4.48 | 12.94 | 97.2 | |
| 100 | 105.91 | 3.52 | 5.86 | 105.9 | ||
| 4 000 | 4642.59 | 6.06 | 13.14 | 112.1 | ||
| Zidovudine-glucuronide | 50 | 51.06 | 6.96 | 8.77 | 102.1 | |
| 100 | 94.2 | 9.44 | 7.38 | 94.2 | ||
| 4 000 | 4143.96 | 6.24 | 8.99 | 103.6 | ||
分别按配制低、高2种浓度代谢产物的标准含药样本的量,分别加入对应各UGTs亚型底物代谢产物的对照品溶液和内标溶液,涡旋混匀,于35 ℃水浴中以氮气流吹干。残渣分别用流动相和空白微粒体提取液复溶后,进行LC-MS/MS分析,计算各UGTs亚型底物代谢产物的基质效应。本方法内标均一化的基质效应因子的RSD值小于14.1%。
2.1.6 提取回收率分别制备含低、中、高3种浓度各UGTs亚型底物代谢产物的标准含药样本;同时按配制低、中、高3种浓度各UGTs亚型底物代谢产物的标准含药样本的量分别加入对应代谢产物的对照品溶液和内标,涡旋混匀,于37 ℃水浴中以氮气流吹干。残渣用空白样本提取液复溶后,进行LC-MS/MS分析。分别计算低、中、高3种浓度的提取回收率。底物代谢产物及内标提取回收率的平均值为89.0%~113.0%。
2.1.7 稳定性分别考察低、中、高3种浓度的各UGTs亚型底物代谢产物的标准含药样本在实验过程中的稳定性。标准含药微粒体样本于室温放置6 h及待测溶液在自动进样器(4 ℃)中放置24 h后,各UGTs亚型底物代谢产物均稳定。
2.1.8 残留效应分别将含各UGTs亚型底物代谢产物浓度为标准曲线最高点的标准含药样本和空白样本交替进样,进行LC-MS/MS分析,记录色谱图,分别记录各UGTs亚型底物代谢物的面积As和内标奥硝唑的峰面积Ai。均无残留。
2.2 人肝微粒体酶活性和阳性对照IC50的测定结果 2.2.1 人肝微粒体酶活性测定本实验条件下,采用探针底物法测定人肝微粒体各亚型除UGT1A3催化鹅去氧胆酸生成鹅去氧胆酸葡萄糖苷酸的Km外,UGT1A1、UGT1A4、UGT1A6、UGT1A9和UGT2B7的酶活性数据见Fig 2。所得人肝微粒体各亚型的酶活性数据基本与文献报道一致,表明所建立的微粒体温孵体系良好,可用于后续研究。

|
| Fig 2 Enzyme kinetics of UGTs substrates in pooled human liver microsomes |
本实验条件下,阳性对照阿扎那韦、雷公藤红素、海柯皂甙元、曲格列酮、尼氟酸、甲酚那酸对相应UGTs酶各亚型均表现出明显的抑制作用(Tab 6), 各亚型阳性对照的抑制曲线见Fig 3。表明本实验建立的微粒体温孵体系可用于抑制作用的评价。
| Enzyme | Substrate | Inhibitor | Km/μmol·L-1 | Vmax/ nmol/min/mg protein | References | IC50/μmol·L-1 | References | ||||
| Result | Literature | Result | Literature | Result | Literature | ||||||
| UGT1A1 | SN-38 | Atazanavir | 1.2±0.19 | 3.3 | 0.018±0.000 65 | 0.021 | [4] | 0.22±0.05 | 0.4~2.5 | [5, 6] | |
| UGT1A3 | Chenodeoxycholic acid | Celastrol | 1 464±150.6 | 63.2~251 | 0.36±0.22 | 0.23~5.2 | [7, 8] | 2.1±0.09 | 0.5~25.2 | [6, 9] | |
| UGT1A4 | Trifluoperazine | Hecogenin | 38.1±4.9 | 61 | 0.746±0.030 7 | 0.965 | [10] | 0.32±0.15 | 0.64~15 | [10] | |
| UGT1A6 | N-acetylserotion | Troglitazone | 2 248±369.7 | 5 200~8 800 | 16.47±1.81 | 0.62~51.3 | [11] | 16.5±0.07 | 28~195.5 | [3, 12] | |
| UGT1A9 | Mycophenolic acid | Niflumic acid | 44±4.0 | 135 | 1.66±0.05 | 13.8 | [13] | 0.48±0.03 | 0.034~8 | [14, 15] | |
| UGT2B7 | Zidovudine | Mefenamic acid | 2 938±230.8 | 1 018 | 3.41±0.18 | 1.54 | [16] | 5.1±0.06 | 0.3~37 | [17, 18] | |

|
| Fig 3 Inhibition curve obtained using individual substrates Each UGT inhibitor was incubated in a separate experiment with an individual substrate set, as described in the "Material and Methods" section. The activity is expressed as the percentage of the remaining activity compared with a control sample containing no inhibitor. Inhibition of (a) SN-38 glucuronidation by atazanavir; (b) chenodeoxycholic acid glucuronidation by celastrol; (c) trifluoperazine glucuronidation by hecogenin; (d) N-acetylserotonin glucuronidation by troglitazone; (e) mycophenolic acid glucuronidation by niflumic acid; and (f) zidovudine glucuronidation by mefenamic acid. |
本研究成功建立了一种可靠且稳定的体外平台,用于评估药物对UGTs体外抑制的潜力。通过选择不同的已知UGTs底物进行评估,证明了该平台对于不同底物具有良好的准确性和灵敏性。同时,通过使用已知的UGTs抑制剂进行验证和优化,进一步确认了该体外平台的可靠性。当前,运用LC-MS/MS技术对人肝微粒体孵育体系中多个典型UGTs酶特异性底物的代谢物进行同时检测的报道仍然较少[6, 9]。本研究首先建立了同时测定人肝微粒体孵育体系中常见UGTs酶UGT1A1、UGT1A3、UGT1A4、UGT1A6、UGT1A9和UGT2B7特异性底物代谢产物的LC-MS/MS方法并对其进行了方法学确证,该方法特异性强,灵敏度高,耗时短。
在开展UGTs酶活性评价前,首先采用单因素法对孵育时间和酶浓度进行了系统优化。分别以孵育时间、肝微粒体蛋白量为横坐标,以UGTs探针底物代谢生成量为纵坐标,选择线性范围内点作为最佳孵育条件。孵育条件的选择应综合考虑酶促反应初始速度和分析方法的灵敏度,并尽可能选择较低的蛋白浓度,以避免底物或抑制剂与蛋白质发生非特异性结合,从而导致结果偏差。本研究所获得的UGT1A3催化的鹅去氧胆酸代谢生成鹅去氧胆酸葡萄糖苷的Km值与文献报道有较大差异(1 464 vs 63.2~251 μmol·L-1)[7-8],鉴于文献报道的Km本身就存在较大的差异,而本研究所获得的Km值亦在可接受的范围内。造成此差异原因可能源于各种因素,包括所选用的合并的肝微粒例数、不同批次、不同来源,以及孵育过程中所选用的底物浓度和孵育条件不同所致。除此之外,本研究所获得的其他UGTs催化对应底物生成代谢产物的酶动力学参数Km和Vmax及IC50值与文献报道值接近[3-6, 9-18],进一步验证了该体外评价体系的准确性和稳定性。在进行酶抑制实验时,可以根据已知的各底物的Km值来选择底物浓度。一般情况下,在保证分析方法灵敏度的前提下,选择小于各底物Km值的浓度作为抑制实验的底物浓度,以减少底物与蛋白的非特异性结合。如果待测药物能对UGTs酶产生抑制作用,可以应用适当的酶促动力学模型来测定可逆性抑制参数IC50和Ki值和/或时间依赖性抑制参数KI和kinact值,通过测定上述参数可以评估药物对代谢酶的抑制力度,抑制机制和药物相互作用[19-20]。
与以往的研究相比,本研究建立的体外平台具有以下优势:首先,本研究选择的UGT底物是常见的底物,具有广泛的临床应用,因此本研究结果更具有实用性和可重复性。其次,本研究使用LC-MS/MS分析技术来准确测定UGT底物的代谢产物,该技术具有高度的灵敏性和准确性,能够提供可靠的代谢数据。最重要的是,我们对平台进行了验证和优化,使用了多个已知的UGT抑制剂来评估平台的准确性和稳定性,确保了平台的可靠性和实用性。
然而,尽管我们所建立的体外评价体系在研究药物对UGT的抑制潜力方面提供了有力的工具,但还有一些限制需要注意:首先,该体外平台仅评估了药物对UGT底物的体外抑制效果,无法完全模拟体内环境,因此在预测体内药物代谢和相互作用时仍需进行进一步的研究。其次,虽然我们使用了多个已知的UGT抑制剂进行验证,但仍有可能存在其他未知的药物抑制剂对平台的影响,因此在实际应用中需要更多的药物相互作用研究。
综上所述,本研究成功建立了一种可靠的体外平台,用于评估药物对UGT的体外抑制效果。该平台具备一定的准确性和灵敏性,并且经过了验证和优化的过程,为药物代谢和相互作用研究提供了有力工具。然而,还需进一步研究来验证其在临床用药指导中的实际应用价值,并加以完善和优化,以提高其在药物研发和临床实践中的可靠性和实用性。
| [1] |
赵世宇, 刘帅兵, 王月琴, 等. m6A修饰对药物代谢酶和药物转运体的调控作用[J]. 中国药理学通报, 2024, 40(7): 1221-25. Zhao S Y, Liu S B, Wang Y Q, et al. m6A modification regulating drug-metabolizing enzyme and drug transporter[J]. Chin Pharm Bull, 2024, 40(7): 1221-1225. doi:10.12360/CPB202311047 |
| [2] |
孙鲁宁, 吴春勇, 赵舜波, 等. 药物对人肝CYP450酶诱导和抑制作用体外评价体系的建立与验证[J]. 药学学报, 2017, 52(12): 1924-32. Sun L N, Wu C Y, Zhao S B, et al. Establishment of in vitro methods for evaluation of induction and inhibition of human CYP450 enzymes by drugs[J]. Acta Pharm Sin, 2017, 52(12): 1924-32. |
| [3] |
Seo K A, Kim H J., Jeong E S, et al. In vitro assay of six UDP-glucuronosyltransferase isoforms in human liver microsomes, using cocktails of probe substrates and liquid chromatography-tandem mass spectrometry[J]. Drug Metab Dispos, 2014, 42(11): 1803-10. doi:10.1124/dmd.114.058818 |
| [4] |
Xiao L L, Zhu W. Li, et al. New Insights into SN-38 Glucuronidation: evidence for the Important Role of UDP Glucuronosyltransferase 1A9[J]. Basic Clin Pharmacol Toxicol, 2018, 122(4): 424-428. doi:10.1111/bcpt.12929 |
| [5] |
Zhang D, Chando D W, Everett, et al. In vitro inhibition of UDP glucuronosyltransferases by atazanavir and other HIV protease inhibitors and the relationship of this property to in vivo bilirubin glucuronidation[J]. Drug Metab Dispos, 2005, 33(11): 1729-1739. doi:10.1124/dmd.105.005447 |
| [6] |
Lee B, Ji H K, Lee T, Liu K H. Simultaneous screening of activities of five cytochrome P450 and four uridine 5'-Diphospho-glucuronosyltransferase enzymes in human liver microsomes using cocktail incubation and liquid chromatography-tandem mass spectrometry[J]. Drug Metab Dispos, 2015, 43(7): 1137-46. doi:10.1124/dmd.114.063016 |
| [7] |
Mullapudi T V R, Ravi P R, Thipparapu G. Thipparapu, UGT1A1 and UGT1A3 activity and inhibition in human liver and intestinal microsomes and a recombinant UGT system under similar assay conditions using selective substrates and inhibitors[J]. Xenobiotica, 2021, 51(11): 1236-46. doi:10.1080/00498254.2021.1998732 |
| [8] |
Chen A, Zhou X, Cheng Y, et al. Design and optimization of the cocktail assay for rapid assessment of the activity of UGT enzymes in human and rat liver microsomes.[J]. Toxicol Lett, 2018, 295: 379-89. doi:10.1016/j.toxlet.2018.07.021 |
| [9] |
Joo J. B, Lee T, Lee, et al. Screening of six UGT enzyme activities in human liver microsomes using liquid chromatography/triple quadrupole mass spectrometry[J]. Rapid Commun Mass Spectrom, 2014, 28(22): 2405-14. doi:10.1002/rcm.7030 |
| [10] |
Uchaipicha V, Mackenzie P I, Elliot D J, et al. Selectivity of substrate (trifluoperazine) and inhibitor (amitriptyline, androsterone, canrenoic acid, hecogenin, phenylbutazone, quinidine, quinine, and sulfinpyrazone) "probes" for human udp-glucuronosyltransferases[J]. Drug Metab Dispos, 2006, 34(3): 449-456. doi:10.1124/dmd.105.007369 |
| [11] |
Krishnaswamy S, Duan S X, Von Moltke L L., et al. Validation of serotonin (5-hydroxtryptamine) as an in vitro substrate probe for human UDP-glucuronosyltransferase (UGT) 1A6[J]. Drug Metab Dispos, 2003, 31(1): 133-139. doi:10.1124/dmd.31.1.133 |
| [12] |
Ito M K, Yamamoto H, Sato, et al. Inhibitory effect of troglitazone on glucuronidation catalyzed by human uridine diphosphate-glucuronosyltransferase 1A6[J]. Eur J Clin Pharmacol, 2001, 56(12): 893-895. doi:10.1007/s002280000252 |
| [13] |
Oda S, Fukami T, Yokoi T, et al. A comprehensive review of UDP-glucuronosyltransferase and esterases for drug development[J]. Drug Metab Pharmacokinet, 2015, 30(1): 30-51. doi:10.1016/j.dmpk.2014.12.001 |
| [14] |
Vietri M A, Pietrabissa F, Mosca, et al. Mycophenolic acid glucuronidation and its inhibition by non-steroidal anti-inflammatory drugs in human liver and kidney[J]. Eur J Clin Pharmacol, 2000, 56(9-10): 659-64. doi:10.1007/s002280000227 |
| [15] |
Mano Y, Usui T, Kamimura H. In vitro inhibitory effects of non-steroidal anti-inflammatory drugs on 4-methylumbelliferone glucuronidation in recombinant human UDP-glucuronosyltransferase 1A9-potent inhibition by niflumic acid[J]. Biopharm Drug Dispos, 2006, 27(1): 1-6. doi:10.1002/bdd.475 |
| [16] |
Bélanger A S, Caron P, Harvey M, et al. Glucuronidation of the antiretroviral drug efavirenz by UGT2B7 and an in vitro investigation of drug-drug interaction with zidovudine[J]. Drug Metab Dispos, 2009, 37(9): 1793-1796. doi:10.1124/dmd.109.027706 |
| [17] |
Mano Y, Usui T, Kamimura H. Inhibitory potential of nonsteroidal anti-inflammatory drugs on UDP-glucuronosyltransferase 2B7 in human liver microsomes[J]. Eur J Clin Pharmacol, 2007, 63(2): 211-6. doi:10.1007/s00228-006-0241-9 |
| [18] |
Knights K M, Winner L K, Elliot D J, et al. Aldosterone glucuronidation by human liver and kidney microsomes and recombinant UDP-glucuronosyltransferases: inhibition by NSAIDs[J]. Br J Clin Pharmacol, 2009, 68(3): 402-12. doi:10.1111/j.1365-2125.2009.03469.x |
| [19] |
Liu S, Wang Z, Chan E, et al. Inhibition of cytochrome P450 enzymes and uridine 5'-diphospho-glucuronosyltransferases by vicagrel in human liver microsomes: a prediction of potential drug-drug interactions[J]. Chem Biol Interact, 2022, 352: 109775. doi:10.1016/j.cbi.2021.109775 |
| [20] |
Hou L, Zhao Y, Zhao S, et al. Ciprofol is primarily glucuronidated by UGT1A9 and predicted not to cause drug-drug interactions with typical substrates of CYP1A2, CYP2B6, and CYP2C19[J]. Chem Biol Interact, 2024, 387: 110811. doi:10.1016/j.cbi.2023.110811 |