



2. 河北医科大学门诊部,河北 石家庄 050017;
3. 河北省中医院药学部,河北 石家庄 050011
2. Dept of Outpatient, Hebei Medical University, Shijiazhuang 050017, China;
3. Dept of Pharmacy, Hebei Provincial Hospital of Traditional Chinese Medicine, Shijiazhuang 050011, China
KCNQ离子通道是电压依赖性钾通道,该通道在神经动作电位的阈值附近激活,激活动力学过程比较慢,激活后不容易失活,调节该通道对神经膜电位和神经兴奋性影响很大[1]。KCNQ 2/3通道主要分布在神经系统,该通道基因突变会引起良性家族性新生儿惊厥综合征[2]、发育性癫痫性脑病[3],部分突变与其功能缺失有密切关系,KCNQ 2/3通道目前已成为抗癫痫新药研发的一个热点方向。迄今已上市的KCNQ离子通道开放剂包括抗癫痫药物瑞替加滨(RTG)和非阿片类镇痛药氟吡汀(Flupirtine),目前正处于临床研究的药物包括上海药物所研发的抗癫痫药物派恩加滨(HN37),Biohaven公司的BHV7000,XEON公司的XEN1101产品等。我们围绕该靶点进行了高通量药物筛选,试图寻找对KCNQ2/3有选择性的高活性化合物,目前发现多个系列具有较好开放活性的先导化合物,希望通过进一步的结构改造,能够找到有治疗潜力的抗癫痫药物。
1 材料 1.1 实验动物SPF级昆明小鼠,SPF级SD大鼠,购自河北医科大学实验动物中心,实验动物生产许可证号:SCXK(冀)2022-001。实验期间动物饲养于屏障环境,使用许可证号:SYXK(冀)2018-005,饲养期间温度20 ℃~25 ℃,明暗12 h交替,实验期间自由进食和水。
1.2 主要试剂戊四唑(pentylenetetrazole,PTZ,货号:L16185)、谷氨酸对照品(glutamic,Glu,货号:A15031)、γ-氨基丁酸对照品(γ-aminobutyric,GABA,货号:A11016)、乙醇胺对照品(货号:A11697),邻苯二甲醛(OPA,货号:A13299)等常规试剂购于Alfa Aesar公司;甲醇(色谱纯,货号:W-50102):Dikma公司;戊巴比妥钠(批号:140418):德国默克生物科技有限公司;QO系列化合物:河北医科大学药理实验室自制。
1.3 主要仪器ICR8000离子通道阅读器:加拿大欧罗拉公司;YSD-4G药理生理实验多用仪:淮北正华生物仪器设备有限公司;DSI生理无线遥测系统:美国DSI;YLS-1C小动物活动记录仪:山东省医学科学院设备站;MD-2200微透析探头(膜长2 mm):美国BAS公司;脑微透析仪:美国BAS公司;Agilent 1100高效液相色谱仪:美国Agilent公司。
2 方法 2.1 铷离子(Rb+)流出高通量筛选方法[4]Rb+与K+有相近的原子大小,并且钾离子通道对于Rb+具有通透性,可以通过检测Rb+流出的浓度来测定钾通道的开放或关闭。Rb+在780 nm有特异的原子吸收,可以通过原子吸收的方法检测Rb+浓度。故可以采用原子吸收光谱测定法,通过测定Rb+流出的高通量测定技术来筛选钾通道的开放剂或关闭剂。技术路线:把稳转了KCNQ2/3通道的CHO细胞铺于96孔板上,以RTG为阳性对照药,所有试药均设100、30、10、3、1、0.3、0.1 μmol·L-1七个浓度梯度,每个浓度设3个复孔进行检测,对定向合成的系列化合物进行筛选,观察Rb+流出浓度,采用logistic方程拟合量效关系曲线,EC50值用x±s表示。
2.2 癫痫动物模型的制备 2.2.1 最大电休克刺激实验(maximal electroshock seizure model,MES)昆明小鼠,体质量18~22 g,实验前24 h进行小鼠筛选。将YSD-4G药理生理实验多用仪设置为单刺激,50 Hz,时间0.25 s,100 V电压(交流输出),以小鼠后肢强直为阳性指标。筛选合格小鼠80只,按体质量随机分成4组,每组20只,雌雄各半。分别为供试品12.5、25、50 mg·kg-1 3个剂量组和溶剂对照组,灌胃给药后1 h,进行电刺激,计算各组抗惊厥发作百分率或保护率(后肢未强直小鼠只数/实验小鼠总只数)。
2.2.2 PTZ致急性癫痫大发作实验昆明小鼠,体质量18~22 g,随机分成4组,每组10只,雌雄各半。分别为供试品小、中、大3个剂量组和溶剂对照组,灌胃给药(12.5、25、50 mg·kg-1)后1 h或腹腔注射给药(6.25、12.5、25 mg·kg-1)后30 min开始用PTZ(85 mg·kg-1)颈背部皮下注射造模,比较各组动物癫痫发作阈值和未发作小鼠的比例(即保护率)。每只小鼠观察30 min,发作的阈值为注射PTZ开始至小鼠第1次出现广泛全身阵挛性发作并丧失翻正反射的时间,如果30 min内动物未发作,阈值按照1 800 s记录。
2.2.3 PTZ慢性点燃癫痫模型实验SD大鼠40只,体质量150~200 g,每日腹腔注射30 mg·kg-1 PTZ造模,连续给药3周,每次药后观察1 h,记录发作级别,癫痫发作参照Racine分级标准[5]。动物发作级别达到Ⅳ级3次及以上时被认为完全点燃,选取发作级别及点燃时间较同步的模型动物,随机分为PTZ模型对照组,供试品12.5、25 mg·kg-1两个剂量组,每组7只,连续腹腔注射给药7 d进行治疗,治疗期间并持续用PTZ点燃,每日观察发作级别和发作持续时间,按照上述评分标准进行评价,控制发作级别为Ⅳ级以下认定为药物有效。
2.3 动物脑电图记录与分析本部分用DSI无线生理信号记录系统记录脑电图(electroencephalogram,EEG)变化,具体操作如下:SD大鼠腹腔注射1%戊巴比妥钠(40 mg·kg-1)麻醉,将DSI无线遥测发射子植入腹腔内,固定于肌肉层。将植入子两根电极由皮下上行至颅骨外,用牙科微型电钻以前囟为标志(后3 mm,左4 mm),(前1.5 mm,右2 mm)颅骨上钻2个小孔,左侧的记录电极暴露硬脑膜,右侧的参比电极放置孔未打穿。将电极头由小孔插入,深度以接触硬脑膜为准,用牙托粉固定。术后连续3 d给予青霉素肌肉注射抗感染。术后恢复1周后进行给药观察。SD大鼠,雄性,体质量200~240 g,20只,随机分4组,每组5只,分别为正常对照组、PTZ模型组、供试品12.5、25 mg·kg-1两个剂量组,连续腹腔注射给药3 d,除正常对照组外其余各组于末次给药后30 min腹腔注射PTZ 50 mg·kg-1造模,观察各组脑电图变化,统计各频率功率谱密度变化。
2.4 一般药理实验 2.4.1 运动协调功能实验昆明小鼠60只,体质量18~22 g,随机分6组,雌雄各半,每组10只。分别为溶剂对照组,供试品设5个剂量组分别为25、50、100、200、400 mg·kg-1。灌胃给药后1 h采用转棒法测定受试动物对小鼠的神经毒性作用。具体操作如下:将小鼠放到小动物疲劳仪的转轴上,转轴直径2.5 cm,速度6 r·min-1,每只观察3 min,以小鼠掉下为阳性指标,记录掉下的小鼠只数,计算阳性率。另取昆明小鼠60只,随机分为6组。分别为溶剂对照组,供试品设定5个剂量组12.5、25、50、75、100 mg·kg-1。腹腔注射给药后30 min放到转棒上进行观察。
2.4.2 自发活动实验昆明小鼠40只,体质量18~22 g,随机分为4组,雌雄各半,每组10只。分别为溶剂对照组,供试品25、50、100 mg·kg-1 3个剂量组。腹腔注射给药后30 min,将小鼠放入底部垫有垫料的自主活动记录仪中适应5 min,分别观察30、60、120、240 min小鼠自主走动次数,每次观察3 min,记录动物活动情况,并与溶剂对照组进行比较。
2.4.3 睡眠协同实验昆明小鼠50只,体质量18~22 g,随机分为5组,雌雄各半,每组10只。分别为溶剂对照组、供试品25、50、100 mg·kg-1 3个剂量组和地西泮阳性对照组,各组均采用腹腔注射给药,给药后30 min各组小鼠均腹腔注射戊巴比妥钠45 mg·kg-1,记录各组动物的睡眠潜伏期及睡眠持续时间。
2.5 脑内神经递质的影响 2.5.1 脑透析液样本采集SD大鼠18只,雄性,体质量200~240 g,随机分为3组,每组6只。腹腔注射1%戊巴比妥钠(40 mg·kg-1)麻醉,暴露颅骨,并以50 μL 0.1%的利多卡因局部麻醉大鼠颅骨,参照大鼠脑立体定位图谱,定位于海马,AP=-5.8 mm,R=5.0 mm,H=-3.0 mm。颅骨上钻一直径为0.65 mm的小孔暴露硬脑膜,植入透析针套管至指定位置,牙科水泥固定。手术后第5天进行透析,之后分别灌胃给予供试品25、50 mg·kg-1两剂量以及溶剂,连续3 d,再次获取给药后的透析液。透析液用95% O2和5% CO2混合气饱和的aCSF,灌流泵以2.0 μL·min-1的速度经微透析探头进行灌流透析,稳定3 h后,以每10 min透析液为一份透析样本收集海马透析样本,收集六份,作为氨基酸的基础释放样本,收集的脑透析液样本置于-80 ℃储存备用。
2.5.2 脑透析样本中氨基酸含量的测定用高效液相色谱法进行检测,衍生化后测量脑内Glu和GABA的含量。取每份脑透析液9 μL,加入内标溶液1 μL,混匀,经自动进样器取含内标的样品溶液5 μL与10 μL衍生化试剂混合,进样测定,记录峰面积,计算脑透析液中各氨基酸的含量,并以灌胃前的氨基酸基础释放值为标准,比较灌胃给药前后脑透析液中各氨基酸含量。色谱条件:流动相为甲醇、乙酸钠(0.1 mol·L-1,用冰醋酸将pH值精确调至5.52),梯度洗脱,流速:1 mL·min-1;荧光检测激发波长:340 nm,发射波长:455 nm[6];柱温:40 ℃。柱前衍生化用5 mg OPA溶于100 μL甲醇中,加入5 μL的2-巯基乙醇,再用硼砂-NaOH缓冲液(pH=9.5)稀释至5 mL,配制成衍生化试剂(含OPA 7.5 mmol·L-1),24 h后使用。
2.6 统计学处理采用SPSS 25统计软件分析处理,采取重复测量的设计,数据以x±s表示,采用单因素方差分析比较各组间差异。计数资料,用卡方(χ2)检验,检验水准α=0.05。
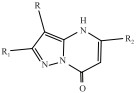
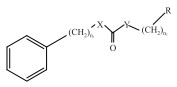
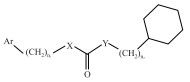
3 结果 3.1 高通量药物筛选结果我们根据文献及合理化药物设计思路,定向设计并合成了吡唑并嘧啶酮(QO系列)、芳香脲类(KG系列)、苯(环己)酰胺类(JM系列)三类共上百个化合物,用Rb+流出高通量筛选方法,在稳转KCNQ2/3通道的CHO细胞系上进行筛选,QO、KG、和JM系列化合物中有活性的代表化合物,见Tab 1,其中QO系列中QO-58、QO-72、QO-82、QO-85,KG系列中KG-7、KG-8、KG-365,JM系列中JM-6、JM-19、JM-23、JM-24、JM-27、JM-28、JM-29均表现出对KCNQ2/3电流良好的开放活性。根据化合物活性高低以及成药5原则的基本参数评价,对QO-72化合物开展了体内抗癫痫活性实验,包括最大电惊厥实验、PTZ诱导的急性及慢性癫痫实验。
| QO series | EC50/μmol·L-1 | KG series | EC50/μmol·L-1 | JM series | EC50μmol·L-1 |
|
|
|
|||
| QO-26 | 2.27±1.02 | KG-1 | 3.80±0.36 | JM-3 | 1.67±0.89 |
| QO-28 | 1.46±0.36 | KG-5 | 3.22±1.35 | JM-4 | 2.41±0.78 |
| QO-40 | 1.25±1.02 | KG-7 | 0.30±0.21 | JM-6 | 0.73±0.12 |
| QO-41 | 4.98±1.86 | KG-8 | 0.14±0.09 | JM-7 | 17.70±2.41 |
| QO-58 | 0.06±0.01 | KG-9 | 1.05±0.08 | JM-9 | 18.40±3.31 |
| QO-60 | 0.15±0.02 | KG-10 | 2.28±1.09 | JM-10 | 3.44±0.47 |
| QO-62 | ~40 | KG-11 | 3.39±0.12 | JM-11 | 0.54±0.15 |
| QO-65 | 7.26±0.23 | KG-14 | 28.9±0.86 | JM-12 | 1.29±0.14 |
| QO-66 | 2.33±1.36 | KG-17 | 13.60±0.12 | JM-14 | 28.10±1.82 |
| QO-68 | 1.93±0.9 | KG-20 | 2.65±0.74 | JM-19 | 0.46±0.14 |
| QO-69 | 6.19±0.59 | KG-22 | 69.47±2.42 | JM-23 | 0.39±0.03 |
| QO-71 | 4.29±0.32 | KG-23 | 3.90±0.51 | JM-24 | 0.75±0.18 |
| QO-72* | 0.60±0.19 | KG-24 | 1.13±0.45 | JM-27 | 0.65±0.09 |
| QO-73 | 0.57±0.24 | KG-25 | 2.29±0.67 | JM-28 | 0.50±0.12 |
| QO-78 | 0.23±0.03 | KG-65 | 0.48±0.06 | JM-29 | 0.44±0.16 |
| QO-82 | 0.58±0.67 | ||||
| QO-83 | 2.28±0.75 | ||||
| QO-85 | 0.16±0.17 | ||||
| QO-89 | 19.3±1.48 | ||||
| QO-91 | 2.06±1.68 | ||||
| QO-94 | 0.21±0.10 | ||||
| *R=Ph,R1=CF3,R2=2,3,4,5-tetrafluoromethyl. | |||||
MES实验是抗癫痫药物筛选最常用的动物模型,具有操作简单,结果客观的特点,且能模拟人类强直阵挛癫痫大发作。QO-72灌胃给药小、中、大3个剂量组(12.5、25、50 mg·kg-1)与溶剂组比较,小鼠最大电休克抗惊厥保护率均有剂量依赖性提高,且重复3次,每次实验数据波动较小,组间差异有显著性(P < 0.01),提示QO-72对小鼠最大电休克致惊厥模型有很好的保护作用,见Fig 1。

|
| Fig 1 Evaluation of QO-72 on MES-induced convulsions in mice (x±s, n=3) **P < 0.01 vs MES control group. |
QO-72分为小、中、大3剂量组,分别灌胃(12.5、25、50 mg·kg-1)和腹腔注射(6.25、12.5、25 mg·kg-1)两种方式给药,与溶剂组比较,灌胃或腹腔注射中、大剂量组均可明显延长发作阈值(P < 0.01),显示出明显保护作用,见Fig 2A、Fig 2C;QO-72灌胃给药的抗惊厥保护率,中、大剂量组分别为40%和70%,与溶剂组比较有明显提高(P < 0.05,P < 0.01),见Fig 2B;腹腔注射给药中、大剂量组的抗惊厥保护率分别达到40%和80%,与溶剂组相比也有明显提高(P < 0.05,P < 0.01),见Fig 2D,表明QO-72灌胃和腹腔注射给药,其抗PTZ致癫痫作用均呈现剂量依赖性。

|
| Fig 2 Evaluation of QO-72 (ig / ip) on PTZ induced convulsions in mice (x±s, n=10) A/C: Seizure threshold was extended by QO-72 (ig / ip) on the convulsions induced by PTZ; B/D: Anticonvulsant protection rate of QO-72 (ig / ip) on the convulsions induced by PTZ. *P < 0.05, **P < 0.01 vs PTZ control group. |
PTZ以30 mg·kg-1剂量注射给药21 d 80%动物点燃成功,可诱发典型Ⅳ级以上的癫痫症状。与模型组动物比较,连续给药7 d,末次给药后观察癫痫发作级别,QO-72小、大两剂量组(12.5、25 mg·kg-1)均可明显降低发作等级(P < 0.01),见Fig 3A。QO-72小、大剂量组(12.5、25 mg·kg-1)分别有2只和5只动物未见明显发作,大剂量组(25 mg·kg-1)的保护率为71.4%与溶剂组相比明显提高(P < 0.01),见Fig 3B。在末次点燃后连续观察1 h内Ⅳ级及以上发作持续时间,QO-72小、大两个剂量组(12.5、25 mg·kg-1)发作的平均持续时间与溶剂组比较均明显缩短(P < 0.01),见Fig 3C。提示,QO-72可以很好的抑制PTZ慢性点燃模型的癫痫发作。

|
| Fig 3 Evaluation of QO-72 on PTZ kindling-induced seizures in rats (x±s, n=7) A: The animals seizures ranks; B: Protection rate; C: The duration of seizures. **P < 0.01 vs PTZ control group. |
用PTZ造模后记录各组大鼠EEG在1 h内的变化,对照组动物未见痫样波发生,见Fig 4A。模型组动物脑电图波幅值明显增大,3~10 min内伴随动物大发作行为,EEG爆发出高于正常脑电图振幅至少2倍以上的连续棘波、棘慢波,大发作后随着动物间断性小发作,不断出现簇状痫样波,见Fig 4B。QO-72小剂量组(12.5 mg·kg-1)给药后EEG棘波、棘慢波明显减少,见Fig 4C;QO-72大剂量组(25 mg·kg-1)的棘波、棘慢波也很少发生,见Fig 4D。将各组EEG图形用NeuroScore软件经傅里叶变换后,比较各剂量组EEG的功率谱值,QO-72小、大剂量组(12.5、25 mg·kg-1)给药后的EEG的功率谱值明显降低,见Fig 4E。接下来进一步分析在各频率段EEG的功率谱密度变化,无论是连续考察各频率段内的功率谱密度,见Fig 4F,还是将Delta (δ)、Theta (θ)、Alpha (α)、Beta (β) 波功率谱密度单独分析,见Fig 4G,均可见明显降低各波段的功率谱密度(P < 0.05,0.01)。提示,QO-72治疗后,由脑电图可见大发作时程明显缩短,振幅明显降低,功率谱密度也明显降低,尤其是大剂量组可明显抑制痫样脑电发生。

|
| Fig 4 Evaluation of EEG of QO-72 on PTZ -induced seizures in rats (x±s, n=5) A: The group of Control; B: The group of PTZ Control; C: The group of QO-72 12.5 mg·kg-1; D: The group of QO-72 25 mg·kg-1; E: Time-frequency analysis of EEG signal power values; F: Power density characteristics of EEG signals; G: Power density of each EEG band. #P < 0.05, ##P < 0.01 vs Control group; *P < 0.05, **P < 0.01 vs PTZ control group. |
转棒实验结果显示,QO-72灌胃后1 h,腹腔注射后30 min,灌胃最高剂量组400 mg·kg-1,腹腔注射最高剂量100 mg·kg-1在观察期内与溶剂对照组比较差异无统计学意义。提示QO-72在治疗剂量灌胃16倍、腹腔注射8倍时,对小鼠协调运动没有明显影响。自发活动实验结果显示,溶剂对照组动物活动正常,给药后120 min内观察未见异常。QO-72 3个剂量组(25、50、100 mg·kg-1) 在给药后的多个观察时间点记录到的自主活动次数与溶剂对照组比较差异均无统计学意义(P>0.05),提示QO-72在抗癫痫药效8倍剂量下对动物自发活动行为没有影响,见Fig 5A。小鼠协同睡眠实验结果显示溶剂对照组小鼠在给药(341±28) s后开始睡眠,见Fig 5B,睡眠持续时间为(3 689±117) s,见Fig 5C。与溶剂对照组相比,QO-72小、中、大三个剂量组(25、50、100 mg·kg-1)对戊巴比妥钠协同睡眠潜伏期和睡眠持续时间均未见明显影响(P>0.05),而阳性对照药地西泮则可明显缩短睡眠潜伏期,延长睡眠持续时间(P < 0.01),见Fig 5B, 5C。结合抗癫痫药效研究数据,提示QO-72在抗癫痫药效8倍剂量下对动物睡眠无影响。

|
| Fig 5 Evaluation of QO-72 on spontaneous activity times at different time(A), sleep latency (B) and sleep duration (C) synergetic with pentobarbital sodium in mice (x±s, n=10). **P < 0.01 vs Control group. |
取标准品Glu、GABA、2-Aminoethanol混合进样分析,测得其保留时间分别为5.8 min、10.8 min、12.2 min。取大鼠空白脑透析液样本加入内标溶液,以及取大鼠灌胃给药后的脑透析液样本进行测试,样品峰和内标完全分离,且脑透析液中内源性物质及其他杂质不影响样品的分离及测定,典型色谱图见Fig 6A。取标准品制备标准曲线Glu在0.2~20 μmol·L-1范围内浓度与峰面积比呈现良好的线性关系,求得回归方程为Ŷ=0.5 368 X-0.0501,(r=0.9989); GABA在0.1~10 μmol·L-1范围内浓度与峰面积比的线性关系良好,回归方程为Ŷ=1.0040X-0.0920,(r=0.9981)。经精密度和准确度验证:Glu日内精密度RSD(n=5)分别为6.38%、5.76%、5.24%,日间精密度RSD(n=5)分别为5.27%、4.95%、2.96%,准确度RE%均小于15%;GABA日内精密度RSD(n=5)分别为9.84%、9.96%、4.86%,日间精密度RSD(n=5)分别为4.65%、5.27%、9.92%,准确度RE%均小于15%。考察样本稳定性结果显示,含10.00 μmol·L-1内标的混合氨基酸对照品在室温放置2 h,4 ℃放置1周和3次冷冻-解冻循环后,3种条件下均稳定,相对误差RE均在±10%之间,RSD < 10%,表明其稳定性良好。脑透析标本中氨基酸含量的变化以灌胃给药前收集的细胞外液氨基酸浓度作为基础释放值进行比较,QO-72按照25、50 mg·kg-1剂量连续给药3 d后脑脊液中抑制性氨基酸GABA的含量明显升高(P < 0.01),兴奋性氨基酸Glu含量未见明显变化(P>0.05),见Fig 6B。结果提示:QO-72具有促进脑内抑制性神经递质GABA释放作用,而对兴奋性氨基酸Glu影响不明显。

|
| Fig 6 Typical chromatogram of neurotransmitters releases in hippocampus cerebrospinal fluid of rats(x±s, n=6) 1:Glu, 2:GABA, 3:2-aminoethanol (A) and evaluation of QO-72 on Glu and GABA levels (B). **P < 0.01 vs Basal. |
KCNQ通道属于电压依赖性钾离子通道亚家族Kv7系列,由5个成员(KCNQ1-5)组成,KCNQ2和KCNQ3广泛分布于神经系统包括海马、大脑皮层、小脑等部位,是构成神经M电流的主要组成部分;KCNQ1主要分布于心脏与KCNE亚基是构成心肌Iks电流的的基础。KCNQ4分布于听神经系统包括内耳毛细胞,该基因突变与迟发渐进性耳聋(DFNA2)密切相关[7]。因此,围绕KCNQ钾通道开展的药物设计成为治疗神经相关疾病的一个热点。根据KCNQ通道的特征,4个亚基围绕成中间离子通道的孔道,每个亚基反复跨膜6次(S1~S6),S1-S4区为电压感受区,S5-S6围绕成内嵌的孔区。在设计药物时主要考虑的结合口袋有两部分[8],一部分是靶向孔区的S5-S6部位,通过变构效应打开通道,并影响通道的电压敏感性,达到开放通道的调节作用,例如,RTG、SF0034、ML213、HN37和RL648_81等。另一策略是靶向通道电压感受区的(S1-S4)区域,通过稳定VSD的激活态构象,达到开放通道的调节作用,例如ICA-069673、ML277、ztz240。但无论是通过调节通道的哪个部分,均表现出使细胞膜电位超极化、降神经兴奋性、抑制异常放电的药理调节作用。QO系列化合物结构新颖,调节通道的机制与上述均不同。
我们筛选出的吡唑[1,5-a]嘧啶-7(4H)-酮QO系列化合物对KCNQ2/3通道选择性高[9],而KG系列与JM系列化合物除了对KCNQ2/3有明显开放作用外,对KCNQ1和其他亚型也表现出明显开放活性,因此我们选择QO系列化合物做进一步研究。在QO系列化合物中QO-58对突变的KCNQ2(W236L)通道也有开放作用[9],表现出良好的镇痛作用[10],但是由于体内药代动力学研究结果显示,该化合物大鼠口服生物利用度低[11],在脑内分布的浓度也不高[12]导致其抗癫痫活性不稳定。在此基础上我们又对QO-58的侧链结构进行改造,得到的QO-72化合物在抗癫痫药效和安全性上均有明显改善,表现出良好的抗癫痫活性,在MES和PTZ两种癫痫模型上,该化合物抗癫痫保护率达到70% ~80%;与一般药理实验研究结果提示QO-72在治疗剂量的16倍(ig.)和8倍(ip.)剂量下没有观察到明显的神经精神及行为异常,说明QO-72相对安全性比较高;通过脑内神经递质分析,提示在该治疗剂量下,QO-72不仅可以通过开放KCNQ通道发挥作用,还可以通过促进脑内GABA类神经递质分泌,提高其中枢抑制性功能,降低EEG的痫样波发放和各波段的功率谱密度。本研究结果提示,QO-72化合物有望开发成一个对急、慢性癫痫发作均有效的抗癫痫类新药。
| [1] |
贾庆忠, 贾占峰, 张英俊, 等. KCNQ2/3钾通道电流特征及M_1受体的调节作用[J]. 中国药理学通报, 2005(10): 56-60. Jia Q Z, Jia Z F, Zhang Y J, et al. Characteristics of KCNQ2/3 potassium channel current and its modulation by M1 receptor[J]. Chin Pharmacol Bull, 2005(10): 56-60. |
| [2] |
Grinton B E, Heron S E, Pelekanos J T, et al. Familial neonatal seizures in 36 families: clinical and genetic features correlate with outcome[J]. Epilepsia, 2015, 56(7): 1071-80. doi:10.1111/epi.13020 |
| [3] |
Kato M, Yamagata T, Kubota M, et al. Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation[J]. Epilepsia, 2013, 54(7): 1282-7. doi:10.1111/epi.12200 |
| [4] |
Wang K, McIlvain B, Tseng E, et al. Validation of an atomic absorption rubidium ion efflux assay for KCNQ/M-channels using the ion channel reader 8000[J]. Assay Drug Dev Technol, 2004, 2(5): 525-34. doi:10.1089/adt.2004.2.525 |
| [5] |
刘睿, 郭翀, 彭江涛, 等. 布洛芬通过下调环氧化酶2降低癫痫大鼠海马神经元兴奋性[J]. 神经解剖学杂志, 2020, 36(4): 383-8. Liu R, Guo C, Peng J T, et al. Ibuprofen reduces the excitability of hippocampal neurons by down-regulating cyclooxygenase 2 in epileptic rats[J]. Chin J Neuro, 2020, 36(4): 383-8. |
| [6] |
王晓菲, 罗晓星, 周四元, 等. RP-HPLC法检测海马脑片灌流液中氨基酸类神经递质[J]. 第四军医大学学报, 2002(5): 426-8. Wang X F, Luo X X, Zhou S Y, et al. Determination of amino acid neurotransmitters in the hippocampal slices perfusate by RP-HPLC[J]. J Fourth Mil Med Univ, 2002(5): 426-8. doi:10.3321/j.issn:1000-2790.2002.05.012 |
| [7] |
Robbins J. KCNQ potassium channels: physiology, pathophysiology, and pharmacology[J]. Pharmacol Ther, 2001, 90(1): 1-19. doi:10.1016/S0163-7258(01)00116-4 |
| [8] |
Kanyo R, Wang CK, Locskai LF, et al. Functional and behavioral signatures of Kv7 activator drug subtypes[J]. Epilepsia, 2020, 61(8): 1678-90. doi:10.1111/epi.16592 |
| [9] |
Zhang F, Mi Y, Qi JL, et al. Modulation of K(v)7 potassium channels by a novel opener pyrazolo[1, 5-a] pyrimidin-7(4H)-one compound QO-58[J]. Br J Pharmacol, 2013, 168(4): 1030-42. doi:10.1111/j.1476-5381.2012.02232.x |
| [10] |
Teng BC, Song Y, Zhang F, et al. Activation of neuronal Kv7/KCNQ/M-channels by the opener QO58-lysine and its anti-nociceptive effects on inflammatory pain in rodents[J]. Acta Pharmacol Sin, 2016, 37(8): 1054-62. doi:10.1038/aps.2016.33 |
| [11] |
刘灿仿, 祁金龙, 张海林, 贾庆忠. 新型钾通道开放剂(QO-58)在大鼠体内药代动力学研究[J]. 中国药理学通报, 2014, 30(4): 574-7. Liu C F, Qi J L, Zhang H L, Jia Q Z. Pharmacokinetic study of QO-58:a new potassium channel opener[J]. Chin Pharmacol Bull, 2014, 30(4): 574-7. doi:10.3969/j.issn.1001-1978.2014.04.029 |
| [12] |
刘灿仿, 祁金龙, 张海林, 贾庆忠. 新型钾通道开放剂(QO-58)在大鼠体内组织分布研究[J]. 中国药理学通报, 2012, 28(1): 66-9. Liu C F, Qi J L, Zhang H L, Jia Q Z. Study of tissue distribution on a new potassium channel opener (QO-58) in rats[J]. Chin Pharmacol Bull, 2012, 28(1): 66-9. doi:10.3969/j.issn.1001-1978.2012.016 |