



2. 新疆医科大学基础医学院 免疫学教研室, 新疆 乌鲁木齐 830011
2. Dept of Immunology, School of Pre-clinical Medicine, Xinjiang Medical University, Urumqi 830011, China
心肌纤维化(myocardial fibrosis,MF)以心肌正常组织结构中细胞增殖、细胞外基质(extracellular matrix, ECM)过度沉积为主要表现[1],是多种心脏疾病发展至一定阶段所共有的病理改变,也是引起心室重塑致心力衰竭的关键原因。醛固酮(aldosterone, ALD)已被证实可以通过多种信号通路致心、肾等器官的纤维化[2]。其能促进转化生长因子β1(transforming growth factor β1, TGF-β1) 等的表达,刺激多种细胞向纤维母细胞的转化及减少基质金属蛋白酶等的合成,促纤维化[3]。炎症反应在心血管疾病进展中越来越受到关注。研究显示[4],多种免疫细胞如巨噬细胞、树突状细胞(dendritic cell,DC)、T细胞、B细胞等均存在醛固酮受体,当其被激动后,会引起多种细胞因子分泌而促进纤维化。调节性T淋巴细胞(regulatory T lymphocytes, Tregs)是近年来发现的抗炎作用细胞,其功能增强被认为可以抑制炎症反应[5]。目前,Treg细胞对心肌纤维化的作用并不清楚。T淋巴细胞的活化/增殖受其膜上的Kv1.3通道的活化调控。前期研究表明[6],慢性心衰大鼠Treg细胞膜上的Kv1.3通道活性增强,推测心衰时Tregs活化/增殖,且给予第2代ALD受体拮抗剂依普利酮(eplerenone, EPL)后,该通道活性明显下调[7]。为进一步阐明Tregs在心肌纤维化进程中的作用,本研究拟建立大鼠Tregs与心肌成纤维细胞(cardiac fibroblasts, CFs)体外共培养模型,评价CFs的变化情况,测定Tregs上Kv1.3等离子通道的改变,并分析探讨EPL对两者的干预机制。
1 材料与方法 1.1 材料 1.1.1 实验动物5周的♂ SD大鼠(新疆医科大学实验动物中心),体质量250~300 g。刚出生的SD乳鼠,母鼠喂养3 d。
1.1.2 试剂大鼠Ⅰ型胶原、大鼠Ⅲ型胶原、大鼠基质金属蛋白酶2(matrix metallopeptidase 2, MMP-2) ELISA检测试剂盒,购自武汉华美生物技术公司;生物素标记的小鼠抗兔CD4 (BD 554836)、PE标记的小鼠抗兔CD25 (BD 554866),购自美国BD公司;大鼠淋巴细胞分离液(LTS1083,Sigma);胎牛血清、RPMI 1640培养基(Hyclone);SYBRTM Select Master Mix(Life Technologies);小鼠抗大鼠KCNN1.3一抗(ab105562)、β-actin(ab8226),购自Abcam公司;IRDye®800CW Goat anti-mouse(LI-COR Biosciences)等。
1.1.3 仪器DOY-CLX Odyssey双色红外激光成像系统(美国LI-COR公司);HF240二氧化碳培养箱(香港Heal Force®公司);KDC-40离心机(中科中佳科学仪器有限公司);ZHWY-200D恒温培养振荡器(上海智成分析仪器制造有限公司);免疫磁珠分选仪MACS(德国Miltenyi)。
1.2 方法 1.2.1 Tregs细胞的分选及体外培养免疫磁珠法分选正常SD大鼠脾脏CD4+CD25+ Tregs(详见相关试剂操作说明书),阳性分选得到Tregs,进行细胞计数及存活率统计。用含10 g·L-1牛血清、rmIL-2(10 mg·L-1)、TGF-β1(2 mg·L-1)、双抗(2 mg·L-1)、anti-CD3(10 mg·L-1)及HSP60(10 mg·L-1)的完全培养基,于37℃、5% CO2孵育48 h待用,本法所得Tregs纯度≥95%。
1.2.2 SD大鼠乳鼠心肌成纤维细胞的分选取新生SD乳鼠10只,胰酶、Ⅱ型胶原酶依次消化分离心肌组织。通过2次差速贴壁分离法收集、培养乳鼠CFs,应用含10%胎牛血清的DMEM于37℃、5% CO2培养箱中孵育。
1.2.3 EPL的浓度对Tregs增殖的抑制作用和共培养的时间对CFs增殖的影响 1.2.3.1 EPL对Tregs和CFs增殖的抑制作用设置EPL梯度浓度(0、0.1、0.3、1、3、10、30、100 μmol·L-1),分别与Tregs或CFs培养48 h后,给予CCK-8试剂检测。抑制率/%=[(A空白对照-AEPL处理组)/A空白对照]×100%。
1.2.3.2 共培养时间对CFs增殖的影响CCK-8法检测Tregs共培养不同时间(0、12、24、36、48、60 h)对CFs增殖的影响。将各组的细胞悬液吸出,用PBS冲洗,每孔加CCK-8试剂后检测。
1.2.4 CFs与Tregs体外共培养接种104个CFs于96孔板中,置于37℃、5% CO2孵育箱中适应性培养48 h,贴壁率达到(80.0±5.0)%时,换PBS空白培养基饥饿培养12 h,同时对Tregs饥饿处理,各组在含有10 g·L-1牛血清、双抗(2 mg·L-1)的RPMI 1640培养基中,于37℃、5% CO2孵育箱中培养48 h。
1.2.5 ECM相关蛋白表达的检测将CFs、CFs+Tregs、CFs+Tregs+EPL 3组细胞于37℃、5% CO2孵育箱中培养48 h后,分别收集培养基,使用ELISA试剂盒检测CFs分泌Ⅰ型胶原、Ⅲ型胶原、MMP-2的相对含量。
1.2.6 Tregs细胞中mRNA的表达检测RT-qPCR检测Tregs细胞的Kv1.3、KCa3.1、CRAC通路基因的表达。收集各组的Tregs,分别提取各组RNA并检测其浓度和纯度,立即进行逆转录以防RNA裂解(详细步骤见Thermo Reverse Translation Kit产品的操作说明书)。使用Life SYBR® Green Master Mix试剂盒进行PCR扩增,于冰上配扩增体系(H2O 6.4 μL,SYBR 10 μL,Primer 0.8 μL),加样品2.0 μL。设定扩增程序Cycle1(1)50.0℃,2 min;Cycle2(1)95.0℃,2 min;Cycle3(40)95.0℃,15 s,60℃,15 s;Cycle4(1)60℃,15 s;Cycle5(1)95℃,15 s。引物序列见Tab 1。
| Gene | Upstream (5′-3′) | Downstream (5′-3′) |
| GAPDH | GGCAGCCTGTTGGAAAAGAA | GGCAGCCTGTTGGAAAAGAA |
| Kv1.3 | GGCAGCCTGTTGGAAAAGAA | GGCAGCCTGTTGGAAAAGAA |
| KCa3.1 | ACTGGAGTCATGGGTGTCTG | ATGAGACTCCTTCCTGCGAG |
| CRAC | TCTCCGTTGAAGAAGACCCC | GGCAGCCTGTTGGAAAAGAA |
将Tregs悬液布于U型黑壁96孔板中,多聚甲醛固定后,用封闭液(0.5 g·L-1脱脂奶粉配制)封闭1 h,加50 μL KCNN1.3一抗(用0.25 g·L-1脱脂奶粉1:800稀释),于摇床上4℃、30 r·h-1过夜,次日加二抗,孵育后,用Odyssey的700和800通道扫描孔板,中等扫描质量,169 μm的分辨率,3.0 mm焦距,亮度为5进行上机检测。
1.2.8 统计学分析运用SPSS 22.0软件对数据进行分析,组间比较采用单因素方差分析。
2 结果 2.1 EPL对Tregs和CFs增殖的抑制作用及共培养时间对CFs增殖的影响 2.1.1 EPL对Tregs和CFs增殖的抑制作用如Fig 1所示,Tregs给予EPL(0.1、0.3、1、3、10、30、100 μmol·L-1) 48 h后,当EPL浓度在0~100 μmol·L-1时,抑制率随EPL浓度的增加而升高,30 μmol·L-1的EPL对Tregs增殖的抑制率达到82.16%,之后趋于稳定,此浓度下药物抑制率接近最大。给予EPL(0.1、0.3、1、3、10、30、100、300 μmol·L-1) 48 h后,CCK-8法检测CFs的增殖。Fig 2结果显示,当EPL浓度在0~300 μmol·L-1时,抑制率随EPL浓度的增加而升高,当EPL的浓度为30 μmol·L-1时,其对CFs增殖的抑制率可达17.17%,之后趋于稳定。基于此,后续实验选择EPL 30 μmol·L-1作为受试检测浓度。

|
Fig 1 Different concentrations of EPL incubated Tregs for 48 hours with different inhibitory rate curve of Tregs proliferation
CCK-8 measured the OD data of Tregs proliferation in different concentrations of EPL. The inhibitory rate curve of Tregs proliferation was con-sistent with the Hill(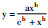 ), R =0.992 2, IC50 =2.9 μmol·L-1. ), R =0.992 2, IC50 =2.9 μmol·L-1.
|

|
|
Fig 2 Different concentrations of EPL incubated CFs for 48 hours with different inhibitory rate curve of CFs proliferation
CCK-8 measured the OD data of CFs proliferation in different concentrations of EPL. The inhibitory rate curve of CFs proliferation was con- sistent with the Hill(), R = 0.997 5, IC50 = 8.5 μmol·L-1.
|
CFs在共培养体系下,细胞增殖明显增加,当向共培养体系中加入EPL(30 μmol·L-1)后,CFs的增殖明显下降。通过CCK-8法检测得Tregs细胞和CFs共培养48 h时达到增殖的平台期,共培养48 h后,CFs+Tregs组的CFs的增殖率为260.35%(CFs+Tregs vs CFs,P<0.01);EPL对CFs的抑制率为59.43%(CFs+Tregs+EPL vs CFs+Tregs,P<0.01),见Tab 2。
| Group | Co-cultured time/h | |||||
| 0 | 12 | 24 | 36 | 48 | 60 | |
| CFs | 0.19±0.00 | 0.63±0.00 | 1.30±0.01 | 2.92±0.02 | 3.91±0.14 | 3.83±0.01 |
| CFs+Tregs | 0.19±0.00 | 1.35±0.05 | 4.81±0.003 | 8.36±0.01 | 10.18±0.03** | 10.11±0.01 |
| CFs+Tregs+EPL | 0.19±0.00 | 0.82±0.02 | 1.26±0.06 | 3.18±0.07 | 4.13±0.03## | 4.15±0.04 |
| **P < 0.01 vs CFs; ##P < 0.01 vs CFs+Tregs | ||||||
ELISA检测结果表明,体系环境中CFs Ⅰ型胶原、Ⅲ型胶原、MMP-2的表达分别增加了2.34、2.46、2.57倍(CFs+Tregs vs CFs,P<0.01)。在培养体系中加入EPL后,Ⅰ型胶原、Ⅲ型胶原和MMP-2的表达被抑制了33.92%、47.97%、62.15%(CFs+Tregs+EPL vs CFs+Tregs,P<0.01),见Tab 3。
| Group | Collagen type Ⅰ /μg·L-1 |
Collagen type Ⅲ /μg·L-1 |
MMP-2 /μg·L-1 |
| CFs | 2.93±0.001 3 | 1.50±0.000 8 | 1.12±0.003 6 |
| CFs+Tregs | 6.87±0.003 4** | 3.69±0.008 4** | 2.88±0.018 0** |
| CFs+Tregs+EPL | 2.33±0.003 2## | 1.77±0.003 6## | 1.79±0.003 7## |
| **P < 0.01 vs CFs group; ##P < 0.01 vs CFs+Tregs group | |||
共培养处理后,Tregs细胞的Kv1.3、KCa3.1、钙释放激活Ca2+(Ca2+ release-activated Ca2+,CRAC)通道的mRNA表达分别增长了6.95、1.99、1.53倍(CFs+Tregs vs Tregs,P<0.01);EPL对Kv1.3通道和KCa3.1通道mRNA表达的抑制率为79.28%、82.14%(CFs+Tregs+EPL vs CFs+Tregs,P<0.01),对CRAC通道mRNA的相对表达抑制率无统计学意义(P>0.05),见Fig 3。

|
| Fig 3 Relative expression level of mRNA in Tregs of different groups **P < 0.01 vs Tregs group; ##P < 0.01 vs CFs+Tregs group |
In-Cell Western blot法检测各组Tregs细胞中Kv1.3通道蛋白的相对表达水平。共培养后,Kv1.3通道蛋白的相对表达水平增加32.05%(P<0.01);EPL能抑制其表达45.68%(P<0.01),见Fig 4。

|
| Fig 4 Relative Kv1.3 channel protein expression level in different groups **P < 0.01 vs Tregs group; ##P < 0.01 vs CFs+Tregs group |
心肌纤维化是慢性心衰进展过程中重要的病理表现[8],它与高血压、心肌梗死、病毒性心肌炎、动脉粥样硬化等多种疾病的病理过程有着密切联系,还可导致心律失常和心源性猝死等严重并发症,故预防和逆转心肌纤维化是临床治疗心力衰竭的关键点之一[9]。心肌纤维化的发病机制至今尚未明确,有观点认为肾素-血管紧张素-醛固酮系统(renin-angiotensin-aldosterone system, RAAS)的激活是引起心肌纤维化的关键[10]。临床上,运用较为广泛的心衰治疗药物是血管紧张素转换酶抑制剂(angiotensin converting enzyme inhibitors, ACEI)类,其对心衰时ALD的释放具有明显的负调控作用[11]。但实验证明,临床上持续应用ACEI至后期,机体对醛固酮的抑制作用会明显降低,即出现了“ALD逃逸”[12]。因此,现今醛固酮受体拮抗剂在心衰的治疗上引起了较大重视。依普利酮是第2代ALD受体拮抗剂,在临床上应用于慢性心衰的治疗,具有选择性高、毒副作用小的特点。前期研究结果显示[6],心衰8周以上的大鼠Tregs的Kv1.3通道电流密度明显增强(是空白组的3~4倍),而ALD受体拮抗剂EPL(30 μmol·L-1)可明显降低Tregs的Kv1.3电流(P<0.01);同样的结果在心衰患者的Tregs中表现更为突出(Kv1.3电流密度为健康人的4~5倍)。提示心衰后期Tregs处于活化增殖状态,并可被依普利酮所抑制。
调节性T细胞是免疫反应和炎症作用平衡中的关键因素[13]。早在1995年,Sakaguchi等[13]就在研究中发现,成年鼠中近10%的外周CD4+T细胞可表达IL-2受体α链CD25的信号表征,去除这群细胞会引起大鼠产生多种自身免疫性疾病,而再次回输该细胞就能阻止疾病的发生。机体免疫系统激活,即免疫调节失衡,则分泌大量的炎症因子,多有报道炎症因子可直接或间接引起心肌纤维化[14]。
本研究的结果表明,EPL对Tregs与CFs共培养48 h时CFs的增殖被抑制趋于稳定;本实验中30 μmol·L-1 EPL对Tregs增殖的抑制率可达到82.16%,对CFs增殖的抑制率为17.17%,可见同一浓度下的EPL对两种细胞抑制作用差别很大,推测EPL对Tregs的抑制作用可能是间接引起CFs增殖被抑制的主要原因。在共培养体系中,CFs合成的Ⅰ型、Ⅲ型胶原和MMP-2的相对表达量均明显增加,给予EPL后,这3种蛋白的相对表达量均明显减少。说明Tregs可促进心肌纤维化,EPL则可能部分通过抑制Tregs的活化/增殖,抑制心肌纤维化进程。
目前,被广泛认可的T细胞活化的信号通路调控为Kv1.3通道维持T淋巴细胞的静息电位,使T细胞处于能被活化的状态;T淋巴细胞被活化后,KCa3.1通道作为Ca2+激活的钾离子通道会使膜电位超极化,使得Ca2+持续内流,促使CRAC通道打开,上调内质网的钙库释放Ca2+的量,Ca2+内流增加可通过钙依赖性的蛋白激酶通路启动各种细胞因子的转录,使其发挥免疫效能。本研究结果显示,CFs+Tregs组的Kv1.3、KCa3.1、CRAC 3种离子通道mRNA的表达较Tregs组均有明显增加,给予EPL后,Kv1.3及KCa3.1通道mRNA的表达明显降低,而CRAC通道的降低趋势差异无统计学意义。Varga等[15]在多发性硬化患者的Tregs上发现Kv1.3通道含量较少。Estes等[16]利用高通量定量方法分析了人初始淋巴细胞的K+通道功能性活动的实验结果,提示Kv1.3通道的活性可作为T细胞的功能活性标志物。基于Kv1.3通道mRNA的研究结果及Kv1.3通道在免疫疾病中靶标的地位,故主要选择对Kv1.3通道蛋白的相对表达水平进行了检测。ICW的结果显示,CFs+Tregs培养体系中,Tregs膜上Kv1.3通道蛋白的表达较Tregs组明显增加,给予EPL后,该通道蛋白的表达被明显抑制。
以上研究结果在mRNA和蛋白的层面上验证了Kv1.3通道在Tregs细胞活化/增殖过程中发挥了重要作用。Tregs上Kv1.3通道的高表达引起Tregs的活化/增殖,进而促进了CFs的增殖,而依普利酮对Kv1.3通道的抑制作用能间接引起CFs的增殖被抑制,虽然依普利酮也能够直接抑制CFs的增殖,但此作用明显弱于间接抑制作用。依普利酮对Kv1.3通道的作用为直接作用还是由于其通过抑制醛固酮受体后的间接作用,仍需进一步研究证实。
( 致谢: 本实验在新疆医科大学第一附属医院科技楼5楼共享实验平台完成,感谢实验平台支持和指导老师的悉心帮助。)
| [1] | Ramos G, Hofmann U, Frantz S. Myocardial fibrosis seen through the lenses of T-cell biology[J]. J Mol Cell Cardiol, 2016, 92: 41-5. doi:10.1016/j.yjmcc.2016.01.018 |
| [2] | Hollenberg N K. Aldosterone in the development and progression of renal injury[J]. Kidney Int, 2004, 66(1): 1-9. doi:10.1111/j.1523-1755.2004.00701.x |
| [3] | Brown N J. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis[J]. Nat Rev Nephrol, 2013, 9(8): 459-69. doi:10.1038/nrneph.2013.110 |
| [4] | Bene N C, Alcaide P, Wortis H H, Jaffe I Z. Mineralocorticoid receptors in immune cells: emerging role in cardiovascular disease[J]. Steroids, 2014, 91: 38-45. doi:10.1016/j.steroids.2014.04.005 |
| [5] | Mohiuddin I H, Pillai V, Baughman E J, et al. Induction of regulatory T-cells from memory T-cells is perturbed during acute exacerbation of multiple sclerosis[J]. Clin Immunol, 2016, 166-167: 12-8. doi:10.1016/j.clim.2016.05.001 |
| [6] | 刘长江, 武洋, 程路峰. 慢性心衰大鼠CD4+CD25+Treg细胞Kv1.3钾通道电流变化[J]. 新疆医科大学学报, 2016, 39(2): 169-78. Liu C J, Wu Y, Cheng L F. The change of Kv1.3 potassium channel current in CD4+CD25+T cells of chronic heart failure rat[J]. J Xinjiang Med Univ, 2016, 39(2): 169-78. |
| [7] | 吴丽敏, 陈立祥, 梁丽娟, 等. 依普利酮下调SGK-1表达抑制梗阻性肾病细胞增殖的研究[J]. 中国药理学通报, 2016, 32(1): 69-73. Wu L M, Chen L X, Liang L J, et al. Inhibitory effects of eplerenone on cell proliferation via down-regulated SGK-1 pathway in rats with unilateral ureteral obstruction[J]. Chin Pharmacol Bull, 2016, 32(1): 69-73. |
| [8] | 秦鑫. TRPM7在压力超负荷下小鼠心肌纤维化形成中的作用[D]. 沈阳: 中国医科大学, 2012. Qin X. The role of TRPM7 in mice myocardial fibrosis induced by pressure overload[D]. Shenyang: China Medical Universty, 2012. http://d.wanfangdata.com.cn/Thesis/Y2266056 |
| [9] | 于瑞, 王幼平, 崔琳, 等. 心肌纤维化的发病机制及其研究进展[J]. 中国现代医生, 2015, 53(13): 157-60. Yu R, Wang Y P, Cui L. Pathogenesy and research advancement of myocardial fibrosis[J]. China Mod Doct, 2015, 53(13): 157-60. |
| [10] | Feldman R D, Gros R. Vascular effects of aldosterone: sorting out the receptors and the ligands[J]. Clin Exp Pharmacol Physiol, 2013, 40(12): 916-21. doi:10.1111/cep.2013.40.issue-12 |
| [11] | Ubaid-Girioli S, Adriana de Souza L, Yugar-Toledo J C, et al. Aldosterone excess or escape: treating resistant hypertension[J]. J Clin Hypertens, 2015, 11: 275-52. |
| [12] | Chimote A A, Kuras Z, Conforti L. Disruption of kv1.3 channel forward vesicular trafficking by hypoxia in human T lymphocytes[J]. J Biol Chem, 2012, 287(3): 2055-67. doi:10.1074/jbc.M111.274209 |
| [13] | Sakaguchi S, Sakaguchi N, Asano M, et al. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases[J]. J Immunol, 1995, 155(3): 1151-64. |
| [14] | 魏伟. 炎症免疫反应软调节[J]. 中国药理学通报, 2016, 32(3): 297-303. Wei W. Soft regulation of inflammatory immune responses[J]. Chin Pharmacol Bull, 2016, 32(3): 297-303. |
| [15] | Varga Z, Csepany T, Papp F, et al. Potassium channel expression in human CD4+ regulatory and naÏve T cells from healthy subjects and multiple sclerosis patients[J]. Immunol Lett, 2009, 124(2): 95-101. doi:10.1016/j.imlet.2009.04.008 |
| [16] | Estes D J, Memarsadeghi S, Lundy S K, et al. High-throughput profiling of ion channel activity in primary human lymphocytes[J]. Anal Chem, 2008, 80(10): 3728-35. doi:10.1021/ac800164v |