



缺血性脑卒中是常见多发的神经系统疾病,治疗的选择性小,致死、致残率高,可由多种原因诱发。目前已有研究表明,在缺血性脑卒中急性期,细胞生物能量代谢障碍引起的细胞去极化、兴奋性神经毒性和氧化应激损伤为主要损害,亚急性期的损害主要是炎症反应,因此造成的神经细胞的坏死和凋亡是产生各种临床症状与体征的根源,死亡的神经元已无法修复,拯救脑缺血半暗带区神经元的凋亡是治疗脑卒中的关键因素[1]。银杏二萜内酯葡胺注射液(YXETNZ)是含银杏内酯B(GB,60%)、银杏内酯A(GA,35%)、银杏内酯K(GK,2%)、银杏内酯C(GC,2%)(Fig1)的5类新药,用于脑卒中的治疗。研究表明银杏二萜内酯各单体除作为是血小板活化因子(platelet activating factor,PAF)的拮抗剂治疗脑卒中外[2],GA和GB可通过抑制线粒体凋亡途径来保护永久性脑缺血大鼠神经元[3],GA和GB还可通过抑制NF-κB活性,降低脑缺血小鼠神经元的炎症和凋亡反应发挥神经保护作用[4, 5],GK通过拮抗急性缺血性脑卒中大鼠神经元的氧化应激保护神经元[6]。本研究采用SH-SY5Y细胞OGD模型模拟脑缺血性损伤,检测YXETNZ对体外脑缺血神经细胞的抗凋亡作用及相关的信号传导通路,进一步阐明了YXETNZ保护脑缺血损伤神经细胞的分子机制。

|
| Fig.1 Chemical structural formula of ginkgolides |
SH-SY5Y购自中科院上海细胞库;RPMI 1640培养基、胎牛血清购自美国Gibco公司;青链霉素混合液购自Biotopped公司;胰蛋白酶购自美国Amresco公司;CCK-8试剂盒购自上海贝博公司;caspase-Glo3/7试剂盒购自美国Promega公司;Cell death detection ELISA购自瑞士Roche公司;p-Akt(Ser473) antibody、Akt(pan) antibody、p-PKA(Thr197) antibody、PKA C-alpha antibody、p-Bad(Ser112) antibody、p-Bad(Ser136) antibody、Bad antibody、actin antibody、HRP标记二抗购自美国CST公司;CO2细胞培养箱购自美国Thermo公司;缺氧小室购自加拿大Stemcell公司;微孔板检测系统Flex Station 3购自美国MD公司;ChemiDoc XRS系统购自美国Bio-Rad公司;银杏二萜内酯葡胺注射液(YXETNZ)为江苏康缘药业生产,产品批号为130901;金纳多注射液(JND)产品批号为H6138。
1.2 方法 1.2.1 细胞培养,OGD模型和给药SH-SY5Y细胞培养于RPMI 1640培养基(含体积分数为10% 胎牛血清,100 kU·L-1青霉素,100 mg·L-1链霉素)中,置于37℃,含5% CO2的细胞培养箱中培养,选取对数生长期细胞进行实验。将SH-SYSY细胞接种至细胞板中培养24 h,以无糖平衡盐溶液完全置换RPMI 1640培养基置于缺氧小室中,将小室用95% N2和5% CO2混合气,以20 L·min-1的气流速度充气20 min以排出空气,密封小室,将小室和对照组细胞置于37℃、5% CO2的细胞培养箱中。将细胞在缺氧小室中培养一定的时间后,取出细胞板置于细胞培养箱中,和YXETNZ、JND药物一起复氧一定时间。然后进行细胞活力、细胞凋亡和相关激酶活性的测定。
1.2.2 细胞存活率的测定将SH-SY5Y细胞以每孔2×104的密度接种至96孔板中,OGD 4 h后加入终浓度为0.39、0.78、1.56、3.12、6.25、12.50、25.00、50.00 mg·L-1的YXETNZ和JND,一起复氧1 h后,采用CCK-8试剂盒,按说明书每孔加入底物10 μL,细胞培养箱中孵育2 h后,于450 nm处测定各组吸光度值OD450 nm,计算药物的保护率(Effection)/%=(药物组细胞存活率-模型组细胞存活率)/模型组细胞存活率×100%。应用GraphPad Prism 5软件计算药物的EC50值,同时确定药物最佳给药剂量。
1.2.3 caspase-3/7活性的测定将SH-SY5Y细胞以每孔2×104的密度接种至96孔底部不透明白板中,OGD 4 h后分别加入终浓度为25 mg·L-1的药物,一起复氧培养1 h,应用caspase-Glo3/7试剂盒,参考说明书每孔加入100 μL的酶作用底物,室温孵育30 min后,用微孔板检测系统Flex Station 3读取各组luminescence值。
1.2.4 细胞质中核小体含量的测定将SH-SY5Y细胞以每孔2×104的密度接种至96孔板中,OGD 4 h后分别加入终浓度为25 mg·L-1的药物,一起复氧培养1 h,应用Cell death detection ELISA试剂盒,参考说明书,测定细胞胞质中核小体的含量。将各组细胞室温裂解30 min后,200×g离心10 min,取上清,与生物素标记的抗组蛋白抗体和HRP标记的抗DNA抗体混合后加入到包被有链霉亲和素的酶标板中,室温震荡孵育2 h,洗涤酶标板,加入显色底物后用微孔板检测系统测定405 nm处各孔的吸光度,计算各组细胞胞质中核小体的含量,浓缩因子(Enrichment factor)=(OD405 nm药物组-OD405 nm空白组)/(OD405 nm对照组-OD405 nm空白组)。
1.2.5 Western blot检测蛋白质水平将SH-SY5Y细胞以每孔1×106的密度接种于6孔板中,收集各组细胞,加入蛋白裂解液提取总蛋白,应用BCA法测定样品蛋白浓度。经SDS-PAGE电泳后,电转蛋白至PVDF膜,封闭,分别加入1 ∶ 1 000稀释的兔抗人的一抗,4℃孵育过夜,TBST洗涤3次,加入HRP标记的羊抗兔二抗(1 ∶ 3 000)溶液室温孵育2 h后洗涤膜,ECL法显影,应用ChemiDoc XRS系统拍照,采用Gel-Pro analyzer 4图像分析软件进行显影条带灰度值分析。
1.2.6 统计学分析所有数据以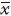 ± s表示,采用GraphPad Prism 5软件进行统计学处理,One-way ANOVA分析比较各组均数之间的差异。
± s表示,采用GraphPad Prism 5软件进行统计学处理,One-way ANOVA分析比较各组均数之间的差异。
应用CCK-8试剂盒测定OGD 1、2、4、6、8、10 h时SH-SY5Y细胞活力的结果显示,与对照组相比,OGD 4 h组细胞存活率降低至54.07%(Fig2A)。随着OGD时间的延长,SH-SY5Y细胞存活率进一步下降,OGD 10 h后,细胞存活率只有18.68%,由结果看出氧糖剥夺可明显造成神经细胞的损伤,SH-SY5Y细胞OGD 4 h为最佳氧糖剥离时间点。将细胞OGD 4 h,加入不同浓度药物一起复氧1 h,结果显示,YXETNZ可明显提高OGD损伤细胞的存活率(Fig2B),且其保护作用随浓度升高而升高,呈浓度依赖性,EC50值分别为2.906、3.251 mg·L-1,同时确定最佳给药浓度为25 mg·L-1。

|
|
Fig.2
Effects of YXETNZ on improvement of the viabilities of SH-SY5Y cells damaged by OGD ( ± s,n=4)
A: The cell viabilities of SH-SY5Y cells damaged by OGD for different times; B: Concentration-dependent improvement of cell viabilities of the OGD-induced SH-SY5Y cells treated with YXETNZ and JND.
|
为了明确YXETNZ对细胞凋亡的抑制作用,本研究检测了细胞凋亡指标caspase-3/7酶活力和细胞质histone-DNA复合小体含量。实验结果显示,与对照组相比,SH-SY5Y细胞OGD损伤后,caspase-3/7酶活力和细胞质中核小体含量明显升高(Fig3A、B),YXETNZ可明显降低caspase-3/7酶活力和抑制细胞核中核小体向细胞质中释放,抑制OGD诱导的SH-SY5Y细胞的凋亡。

|
|
Fig.3
Effects of YXETNZ on inhibition of apoptosis of SH-SY5Y cells induced by OGD ( ± s,n=3)
A: The effects of YXETNZ on inhibition of caspase-3/7 activities of SH-SY5Y cells induced by OGD; B: The effects of YXETNZ on decrease of the levels of cytoplasmic histone-associated-DNA-fragments of SH-SY5Y cells induced by OGD. ##P<0.01 vs control group; **P<0.01 vs OGD group
|
Akt信号通路是对神经细胞有保护作用的信号通路,本研究检测了YXETNZ对此信号通路的影响。蛋白质分析结果显示,与OGD组相比,YXETNZ组磷酸化的Akt(Ser473)蛋白质水平及下游磷酸化的Bad(Ser136)蛋白质水平明显升高,各组Akt总蛋白质和Bad总蛋白质无差异(Fig4)。说明YXETNZ能通过活化Akt通路保护神经细胞,并通过磷酸化下游的Bad蛋白质抑制其活性,抑制细胞凋亡。

|
|
Fig.4
Protein levels of the OGD-induced SH-SY5Y cells treated with YXETNZ ( ± s,n=3)
A: Protein levels of p-Akt(Ser473) and p-Bad(ser136); B-C: The protein levels quantified by band gray-value ratio to actin; B: p-Akt(Ser473); C: p-Bad(Ser136). **P<0.01 vs OGD group.
|
本研究检测了YXETNZ对PKA信号通路的影响。蛋白质分析结果显示,与OGD组相比,YXETNZ组中磷酸化PKA(Thr197)及PKA下游的磷酸化的Bad(Ser112)蛋白质水平明显升高,各组PKA总蛋白质和Bad总蛋白质无差异(Fig5)。说明YXETNZ能通过活化PKA信号通路抑制凋亡保护神经细胞。

|
|
Fig.5
Protein levels of the OGD-induced SH-SY5Y cells treated with YXETNZ ( ± s,n=3)
A: Protein levels of p-PKA(Thr197) and p-Bad(Ser112); B-C: The protein levels quantified by band gray-value ratio to actin; B: p-PKA(Thr197); C: p-Bad(Ser112). *P<0.05 , **P<0.01 vs OGD group.
|
缺血性脑卒中是常见多发的神经系统性疾病,约占脑卒中的60%~80%[7],是世界范围内危害人类健康的重要疾病之一。SH-SY5Y细胞的OGD模型是模拟缺血性脑卒中的经典模型,已成为离体水平研究脑缺血损伤机制及药物作用的重要手段。SH-SY5Y细胞OGD损伤后,细胞活性明显降低,细胞凋亡水平明显增加。YXETNZ可浓度依赖性地提高OGD损伤神经细胞的活性,明显降低OGD神经细胞的凋亡。JND是以银杏萜类内酯、银杏黄酮类即银杏叶提取物为主要成分的注射液,虽然银杏黄酮可抑制脑缺血细胞凋亡[8],但对于PI3K/Akt/Bad、PKA/Bad通路的活化,YXETNZ作用明显优于JND,这可能与其银杏二萜内酯含量高于JND有关。
脑缺血神经细胞凋亡主要通过外源性死亡受体通路、内源性线粒体通路和内质网通路进行信号传导[9]。当神经细胞发生氧糖剥夺时,线粒体ATP能量代谢障碍,线粒体膜通透性增加,导致细胞色素C由线粒体内膜释放入细胞质中[10],并与Apaf-1、procaspase-9蛋白形成复合物,此复合物促使procaspase-9从没有活性的酶原转变成活化状态[11],随后caspase-9活化caspase-3执行凋亡程序。研究表明Bad、Bcl-2、Bcl-xL等凋亡相关Bcl-2家族蛋白在脑缺血线粒体凋亡信号传导中发挥重要作用[9]。当神经细胞发生缺血损伤时,Bad由磷酸化的无活性状态去磷酸化激活,并与14-3-3分子伴侣蛋白解离转移到线粒体外膜上,与Bcl-2、Bcl-xL结合抑制其的抗凋亡活性,促进线粒体膜去极化,增强膜的通透性,促进细胞色素C的释放诱导细胞凋亡[12]。当Bad的Ser112、Ser136或Ser155残基磷酸化时,就能与14-3-3蛋白形成复合物,抑制Bad与Bcl-2/Bcl-xL结合,促进细胞存活[13, 14]。
PI3K/Akt通路和PKA通路是机体内调控细胞存活、凋亡、生长等多种生理功能的激酶传导通路。研究报道活化的PI3K/Akt、PKA通路均可通过磷酸化Bad抑制脑卒中神经细胞的凋亡损伤[9, 15],Akt磷酸化Bad的Ser136残基,PKA磷酸化Bad的Ser112和Ser155残基[13, 16, 17, 18]。由本研究结果看出,银杏二萜内酯葡胺注射液可磷酸化激活OGD损伤SH-SY5Y细胞Akt和PKA激酶,分别促进Bad (Ser136)、Bad(Ser112)磷酸化,通过调控线粒体凋亡途径下调caspase-3/7酶活力促进在脑缺血条件下神经细胞的存活。银杏二萜内酯葡胺注射液激活PI3K/Akt/Bad/caspase-3/7和cAMP/PKA/Bad/caspase-3/7通路是其除了通过PAF受体拮抗抑制血栓形成之外,在缺血性脑卒中治疗过程中发挥作用的重要机制之一。
| [1] | Fann D Y, Lee S Y, Manzanero S, et al. Pathogenesis of acute stroke and the role of inflammasomes [J]. Ageing Res Rev, 2013, 12(4):941-66. |
| [2] | Koch E. Inhibition of platelet activating factor (PAF)-induced aggregation of human thrombocytes by ginkgolides:considerations on possible bleeding complications after oral intake of Ginkgo biloba extracts [J]. Phytomedicine, 2005, 12(1-2):10-6. |
| [3] | Wang X, Jiang C M, Wan H Y, et al. Neuroprotection against permanent focal cerebral ischemia by ginkgolides A and B is associated with obstruction of the mitochondrial apoptotic pathway via inhibition of c-Jun N-terminal kinase in rats [J]. J Neurosci Res, 2014, 92(2):232-42. |
| [4] | Gu J H, Ge J B, Li M, et al. Inhibition of NF-kappaB activation is associated with anti-inflammatory and anti-apoptotic effects of Ginkgolide B in a mouse model of cerebral ischemia/reperfusion injury [J]. Eur J Pharm Sci, 2012, 47(4):652-60. |
| [5] | 葛建彬, 顾锦华, 李 梅, 等. 银杏内酯A对小鼠脑缺血/再灌注损伤的保护作用及其抑制 NF-κB信号通路下调p53、Caspase-3表达的机制 [J].中国药理学通报, 2012, 28(8):1105-10. Ge J B, Gu J H, Li M, et al. Neuroprotective effects of gikgolide A on a mouse model of transient focal cerebral ischemia associated with inhibition of NF-κB signaling pathway and down-regulation of the levels of p53 and Caspase-3 [J]. Chin Pharmacol Bull, 2012, 28(8):1105-10. |
| [6] | Ma S, Yin H, Chen L, et al. Neuroprotective effect of ginkgolide K against acute ischemic stroke on middle cerebral ischemia occlusion in rats [J]. J Nat Med, 2012, 66(1):25-31. |
| [7] | Agudo-Lopez A, Miguel B G, Fernandez I, et al. Involvement of mitochondria on neuroprotective effect of sphingosine-1-phosphate in cell death in an in vitro model of brain ischemia [J]. Neurosci Lett, 2010, 470(2):130-3. |
| [8] | 王海波, 殷淑兰, 林 静, 等. 鼠脑缺血后再灌注 c-fos 基因表达及银杏黄酮的影响 [J]. 中华老年心脑血管病杂志, 2005, 2(5):339-42. Wang H B, Yin S L, Lin J, et al. Study on the effects of EGB on the expression of c-fos gene in rat brain after ischemia and reperfusion [J]. Chin J Geriatr Cardiov Cerebrov Dis, 2005,2(5):339-42. |
| [9] | Nakka V P, Gusain A, Mehta S L, et al. Molecular mechanisms of apoptosis in cerebral ischemia:multiple neuroprotective opportunities [J]. Mol Neurobiol, 2008, 37(1):7-38. |
| [10] | Sugawara T, Fujimura M, Morita-Fujimura Y, et al. Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia [J]. J Neurosci, 1999, 19(22):RC39. |
| [11] | Krajewski S, Krajewska M, Ellerby L M, et al. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia [J]. Proc Natl Acad Sci USA, 1999, 96(10):5752-7. |
| [12] | Zha J, Harada H, Yang E, et al. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) [J]. Cell, 1996, 87(4):619-28. |
| [13] | Yang X, Liu L, Sternberg D, et al. The FLT3 internal tandem duplication mutation prevents apoptosis in interleukin-3-deprived BaF3 cells due to protein kinase A and ribosomal S6 kinase 1-mediated BAD phosphorylation at serine 112 [J]. Cancer Res, 2005, 65(16):7338-47. |
| [14] | Yin H, Chao L,Chao J. Kallikrein/kinin protects against myocardial apoptosis after ischemia/reperfusion via Akt-glycogen synthase kinase-3 and Akt-Bad.14-3-3 signaling pathways [J]. J Biol Chem, 2005, 280(9):8022-30. |
| [15] | Chan P H. Mitochondrial dysfunction and oxidative stress as determinants of cell death/survival in stroke [J]. Ann N Y Acad Sci, 2005, 1042:203-9. |
| [16] | Datta S R, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery [J]. Cell, 1997, 91(2):231-41. |
| [17] | Datta S R, Brunet A,Greenberg M E. Cellular survival:a play in three Akts [J]. Genes Dev, 1999, 13(22):2905-27. |
| [18] | Harada H, Becknell B, Wilm M, et al. Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A [J]. Mol Cell, 1999, 3(4):413-22. |