 2016, Vol. 36
2016, Vol. 36文章信息
- 李梦悦, 王腾飞, 汪俊卿, 赵一瑾, 程成, 王瑞明
- LI Meng-yue, WANG Teng-fei, WANG Jun-qing, ZHAO Yi-jin, CHENG Cheng, WANG Rui-ming
- 海藻糖合酶在毕赤酵母表面的展示
- Expression of Trehalose Synthase Gene in Pichia pastoris
- 中国生物工程杂志, 2016, 36(2): 73-80
- China Biotechnology, 2016, 36(2): 73-80
- http://dx.doi.org/10.13523/j.cb.20160211
-
文章历史
- 收稿日期: 2015-09-15
- 修回日期: 2015-09-24






海藻糖广泛分布于细菌、藻类、酵母、低等植物、昆虫和其它无脊椎动物中,是目前开发的主要新型天然糖之一。海藻糖可以在细胞处于饥饿、干燥、高温、高渗透压及有毒试剂等胁迫环境时,有效地维持细胞内质膜和蛋白质的稳定[1],在保护基因工程酶类、各种病毒、疫苗、抗体、蛋白质因子、核酸等方面取得令人振奋的结果。
海藻糖制取上存在困难,因此该糖价格较高,且生产量低,远不能满足市场需求。各国科学家一致认为生产海藻糖的经济方法是利用海藻糖合酶转化廉价的麦芽糖底物一步生成海藻糖,但近十年来产海藻糖合酶(trehalose synthase,Tres)菌株难以实现发酵生产海藻糖的经济目标。本实验室筛选的Pseudomonas putide P06菌株,其所产海藻糖合酶,温度和pH耐受性强,在50 ℃和pH 8.5~9.0时,酶活最高,在底物浓度40%时,底物转化率高达65%,在该条件下反应,不仅可以避免杂菌污染,而且从工业化角度,可以实现节能降耗,但该菌产酶少,难以用于工业化。目前,来自Pseudomonas putide P06的海藻糖合酶基因实现了在原核系统中的成功表达,但原核表达系统经常出现质粒丢失,构建的菌株极不稳定;在工业化生产中因其存在破碎困难,且产有内毒素,使海藻糖合酶的生产受到限制。该真核表达系统具有乙醇氧化酶基因诱导性强启动子,甲醇诱导重组菌步骤简单,成本低廉,有利于工业化生产。外源基因是以基因重组的方式与毕赤酵母基因组发生整合,因此外源基因在毕赤酵母中非常稳定[2],有利于蛋白的表达[3]。且该系统不产生内毒素,更适合用于食品及医药品的生产[4]。毕赤酵母是近年来发展迅速、应用广泛的一种真核表达系统[5]。具有易培养、繁殖快的优点[6],毕赤酵母具有乙醇氧化酶基因诱导性强启动子且存在翻译后修饰[7, 8, 9, 10],可调控外源基因的表达,便于基因工程操作和高密度发酵,已成为微生物表面展示系统极有应用前景的宿主菌。酵母表面展示可以对外源蛋白进行折叠,糖基化修饰,利用酵母细胞内的蛋白转运机制可以将靶蛋白表达并定位到酵母细胞表面,表达的蛋白具有独立的空间构像。本研究将海藻糖合酶(Tres)的C端和来自酿酒酵母的Pir1p的N端融合[11],与质粒pPICZαA连接,电转化进毕赤酵母GS115中,利用α-factor信号肽[12]将蛋白引导分泌至细胞壁展示于毕赤酵母表面[9, 13],使海藻糖合酶表面展示于毕赤酵母细胞表面。通过β-1,3-葡聚糖苷键共价连接细胞壁的Pir型细胞壁甘露糖可被昆布多糖酶作用[14, 15]。本实验首次利用毕赤酵母表面展示系统展示海藻糖合酶,成功获得了海藻糖合酶的全细胞催化剂,并探索了毕赤酵母表面展示海藻糖合酶生产海藻糖的可行性。
1 材料与方法 1.1 材 料 1.1.1 菌株与质粒E.coli DH5α(含pPICZαA质粒)为本实验室保存。感受态细胞E.coli DH5α为目的基因克隆宿主菌,购自艾德莱生物有限公司。Pseudomonas putide P06为tres的来源菌株,为本实验室保存。酿酒酵母(Saccharomyces cerevisiae) 32919为pir1p来源菌株,购自Invitrogen公司。重组质粒表达宿主菌毕赤酵母 GS115(Pichia pastoris GS115)购自Invitrogen公司。零背景平末端pZERO-Blunt-T Vector阳性克隆筛选载体购自艾德莱生物有限公司。表达载体质粒pPICZαA,含有博来霉素(Zeocin)抗性标签,购自Invitrogen公司。
1.1.2 试剂及培养基Hifi酶、DNA连接酶、限制酶均购自大连宝生物工程公司;DNA柱纯化试剂盒购自生工生物工程(上海)有限公司;DNA标准分子量、质粒提取试剂盒以及T载体购于北京全式金公司;LB培养基、YPD培养基均按照Invitrogen公司毕赤酵母表达手册配制;其他试剂为国产和进口分析纯试剂。LB培养基、YPD培养基均按照Invitrogen公司毕赤酵母表达手册配制。
1.1.3 仪 器Bio-Rad Gene PulserⅡ电转化仪,日本岛津GCMS-QP2010液相检测仪。
1.1.4 引 物细菌基因组DNA提取试剂盒提取Pseudomonas putide P06菌株基因组DNA,并以此为模板克隆目的基因tres(GenBank 序列号为26986745)。设计用于扩增的寡核苷酸引物命名为F1,R1。如表 1所示,F1中下划线部分为EcoR I限制性内切酶酶切位点,波浪线部分为Xba I限制性内切酶酶切位点,R1中下划线部分则为Spe I限制性内切酶酶切位点。
| Primer name | Primer sequence(5′~3′) | Restriction site |
| Forward(F1) | CCGGAATTCATGACCCAGCCCGACCCGTC | EcoR I |
| Reverse(R1) | GCTCTAGAA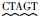 AACATGCCCGCTGCTGTTGAC AACATGCCCGCTGCTGTTGAC | Xba I、Spe I |
| Forward(F2) | GGACTAGTCAGAGAGCCGCTGCTAT | Spe I |
| Reverse(R2) | GCTCTAGATTAACAGTTGAGCAAATCGAT | XbaⅠ |
酵母基因组提取试剂盒提取酿酒酵母基因组DNA,并以此为模板为克隆目的基因pir1p(GenBank 序列号为D13740)。设计用于扩增的寡核苷酸引物命名为F2,R2。如表 1所示,F2中下划线部分为Spe I限制性内切酶酶切位点,R2中下划线部分为Xba I限制性内切酶酶切位点。引物由上海生工生物工程有限公司合成。
1.2 方 法 1.2.1 tres基因,pir1p基因的获得提取Pseudomonas putide P06菌株基因组DNA,以基因组DNA作模板,F1,R1为引物,PCR扩增tres片段。反应条件:95℃ 5 min;95 ℃ 30 s;60 ℃ 30 s;72 ℃ 3 min30 s;30个循环;最后72 ℃延伸10 min。反应结束后以1%的琼脂糖凝胶电泳检测产物并进行柱回收。
提取酿酒酵母基因组DNA,以基因组DNA作为模板,F2,R2为引物,PCR扩增pir1p 片段。反应条件:95 ℃ 5 min;95 ℃ 30 s;55 ℃ 30 s;72 ℃ 2 min;30个循环;最后72 ℃延伸10 min。反应结束后以1%的琼脂糖凝胶电泳检测产物并进行柱回收。
1.2.2 构建重组 T 载体 pZERO-Blunt-tres和pZERO-Blunt-pir1p将 pZERO-BluntT 载体分别与tres PCR 反应所得基因产物及pir1p PCR 反应所得基因产物按照一定比例连接得到重组 T 载体后转化大肠杆菌 DH5α 感受态细胞,并将其涂布于LB平板,37 ℃恒温箱隔夜培养,筛选克隆子,用灭菌牙签挑取单克隆菌落,pZERO-Blunt-tres以F1、R1作上、下游引物,pZERO-Blunt-pir1p以F2、R2作上、下游引物,进行菌落PCR初步验证。将初步筛选出的阳性克隆子按对应序号在LB培养基中扩培,提取重组T载体,送至上海生工公司测序验证。
1.2.3 重组质粒 pPICZαA-tres的构建摇菌并提取pPICZαA质粒,以及测序成功的pZERO-Blunt-tres重组T载体,对二者分别进行EcoR I和Xba I双酶切反应,柱回收酶切的pPICZαA质粒,胶回收tres基因片段,按照一定比例连接得到重组质粒pPICZαA-tres后转化大肠杆菌DH5α感受态细胞,按照 1.2.2 节中重组T载体的操作方法对其进行初步筛选。将初步筛选出的阳性克隆子按对应序号在不同浓度Zeocin抗性低盐LB培养基中扩培,提取重组pPICZαA-tres载体,EcoRI/XbaI酶切二次鉴定,然后将通过验证的重组载体送至上海生工公司测序验证。
1.2.4 重组质粒 pPICZαA-tres-pir1p的构建摇菌并提取质粒pZERO-Blunt-pir1p,经Spe I/ Xba I酶切获得pir1p基因片段,与经同样双酶切pPICZαA-tres重组质粒连接,转入大肠杆菌DH5α感受态细胞中按照 1.2.3 节中筛选重组子的方法进行筛选。进一步对所构建的重组质粒pPICZαA-tres-pir1p进行三酶切EcoR I/Spe I/ Xba I验证,确保重组质粒构建成功以及所携目的基因的正确性。整个构建过程如图 1所示。

|
| 图 1 重组质粒 pPICZαA-tres-pir1p的构建 Fig. 1 Flow chart of recombinant plasmid construction |
将提纯的 pPICZαA-tres-pir1p质粒用Pme I限制酶单切线性化后,电击转化至毕赤酵母GS115感受态细胞,涂布于含有120 μg/ml Zeocin的YPDS平板,30 ℃培 4~6 d,筛选阳性转化子。同浓度 Zeocin YPD培养基扩培后提取重组菌的基因组,分别以F1、R1和F2、R2为引物进行PCR鉴定。
1.2.6 外源蛋白的诱导表达挑取阳性重组子,接种至含50 ml BMGY(pH6.0)培养基的500 ml三角瓶中,30 ℃、250 r/min振荡培养 20 h;静置2h,弃上清,再以50 ml BMMY(pH7.5) 重悬菌体;25 ℃、250 r/min振荡培养,诱导蛋白表达;每24 h向培养基中添加100%甲醇使其终浓度为0.5%,诱导72 h,收集发酵物。取少量发酵产物离心,收集发酵上清液,备用;将发酵既得菌体用生理盐水洗涤3次,PBS缓冲液重悬,收集备用;将一部分洗涤后的菌体,用PBS缓冲液重悬细胞并进行超高压破碎,6 000 r/min离心5 min,收集菌体细胞内容物,备用;破碎后的菌体沉淀生理盐水洗涤3次,PBS缓冲液重悬,使用昆布多糖酶酶解洗脱得到Pir1p-Tres融合蛋白,具体方法见文献[9]。同时取转化有空载体的毕赤酵母对照菌进行相同发酵条件及后期处理,以作空白对照。用SDS-PAGE法分别对发酵上清液、昆布多糖酶水解的融合蛋白以及菌体细胞内容物进行蛋白检测。
1.2.7 外源蛋白活性的检测取一干净50 ml离心管,将5 ml重组菌发酵液上清,5 ml重组菌菌体沉淀重悬液,5 ml破碎后重组菌菌体内容物分别和10 ml pH 7.5的磷酸盐缓冲液配制的终浓度30%的麦芽糖浆混匀后制备成海藻糖合酶的反应体系。将此反应体系置于50℃恒温水浴震荡摇床中反应2 h,反应结束后,80 ℃水浴20 min终止酶的转化反应。室温,1 000 r/min离心10 min,取上清进行HPLC检测转化体系中海藻糖含量。转化有空载体的毕赤酵母经相同方法处理之后做空白对照。HPLC测定方法如表 2所示。
| Liquid phase system | SHIMADZU LC-20A |
| Chromatographic column | Amino bonded phase colμMn(5μmol/L,4.6×250 mm) |
| Mobile phase | V(Acetonitrile)∶V(H2O)=75∶25 |
| Current Speed | 1ml/min |
| ColμMn temperature | 40 ℃ |
| Detector | Differential refraction detector |
| Sample quantity | 10 μl |
酶活力单位定义:在pH 7.5条件下,每毫克海藻糖合酶每小时催化转化30% 麦芽糖溶液产生1 g海藻糖的酶量为1单位(U/g)。
1.2.9 表面展示海藻糖酶最适反应温度及温度耐受性测定按照 1.2.7 中的方法将发酵诱导得到的菌体沉淀,在不同反应温度条件(30 ℃~60 ℃)下测定表面展示海藻糖合酶酶活,以了解其最适反应温度。为了评估表面展示海藻糖合酶的温度耐受性,选取最适反应温度左右范围内的不同温度值,将粗酶液水浴保温10 min,20 min,30 min,40 min,50 min,60 min后迅速降至室温测定酶活,以未保温粗酶液酶活为100%,研究表面展示海藻糖合酶的热稳定性。
1.2.10 表面展示海藻糖酶最适反应pH及pH耐受性测定改变反应体系缓冲液的pH,在不同的pH(5.5~8.5)条件下测定表面展示海藻糖合酶酶活,以了解其最适反应pH。pH耐受性实验选取最适反应pH左右范围内的不同pH于最适反应温度条件下处理60 min后测定酶活,以最适pH条件下所测酶活为100%,研究表面展示海藻糖合酶的酸碱稳定性。
2 结果与分析 2.1 目的基因扩增以Pseudomonas putide P06菌株基因组DNA为模板,由PCR扩增得到大小约为2 100 bp左右的目的片段,大小与海藻糖合酶基因tres预测值相符(如图 2所示),说明目的基因克隆成功。以酿酒酵母基因组DNA为模板,由PCR扩增得到大小约为850 bp左右的目的片段,大小与pir1p基因预测值相符,说明目的基因克隆成功。

|
| 图 2 目的基因PCR 电泳 Fig. 2 PCR results of target gene (a)1: PCR results for the purpose of tres gene; M: Trans 5K DNA Marker (b) 1: PCR results for the purpose of pir1p gene; M: Trans2K plus DNA Marker |
海藻糖合酶基因tres扩增片段以及pir1p基因扩增片段插入T载体。经菌落 PCR,对重组T载体酶切验证及海藻糖合酶目的片段经DNA测序后,将测序结果经BLAST比对分析,结果显示PCR扩增所得基因片段序列与Pseudomonas putide P06中tres目的基因序列相似度达到100%,未发生碱基突变或丢失,表明目标基因tres扩增获得成功。pir1p目标片段经DNA测序比对后,结果显示PCR扩增所得基因片段序列与酿酒酵母中pir1p目的基因序列相似度达到100%,未发生碱基突变或丢失,表明目标基因 pir1p扩增获得成功。提取重组pZERO-Blunt-tres载体,EcoR I/Xba I酶切,得到tres片段,将其连接至经EcoR I/Xba I酶切的表达载体pPICZαA构建成重组质粒pPICZαA-tres。然后提取重组pZERO-Blunt-pir1p载体,Spe I/Xba I酶切,得到pir1p片段,将其连接至经Spe I/Xba I酶切的表达载体pPICZαA-tres构建成重组质粒pPICZαA-tres-pir1p。
2.3 重组质粒的转化和鉴定将构建完成并测序无误的重组质粒通过电转化法整合到毕赤酵母GS115基因组中,从抗性平板上挑取阳性菌落,经 YPD 液体培养基摇瓶培养48h,提取酵母基因组,以F1、F2和F3、F4为引物进行 PCR 鉴定,证明阳性转化子基因组为模板成功克隆出tres、pir1p基因,重组质粒整合进入毕赤酵母基因组,重组菌构建成功,命名为Pichia pastoris-tres-pir1p。将重组质粒经EcoR I/Spe I/Xba I 3种酶酶切验证之后得到3段片段,分别是大小约为3 500 bp左右的pPICZαA片段,大小为2 100 bp左右的tres片段,大小为850 bp左右的pir1p片段。三酶切之后的电泳结果如图 3所示。

|
| 图 3 重组质粒 pPICZαA-tres-pir1p 片段的三酶切验证 Fig. 3 esults of the pPICZαA-tres-pir1p recombinant plasmid restriction enzyme analysis M:Trans5K DNA Marker; 1: Triple digestion of recombinant plasmid pPICZαA-tres-pir1p |
SDS聚丙烯酰胺凝胶电泳对重组菌与转入空质粒的毕赤酵母的发酵液经离心之后的上清液检测,结果没有发现差异蛋白(图 4中1、2泳道所示),说明重组菌发酵液上清中并没有融合蛋白分泌至胞外或表面展示的外源蛋白脱落现象。对重组菌和转入空质粒的毕赤酵母的细胞壁使用昆布多糖酶进行抽提,洗脱的产物中发现重组菌多出一条大小约为108 kDa的蛋白(图 4中4泳道所示),与融合蛋白预期的相对分子质量大小相符,说明表面展示的海藻糖合酶与Pir1p蛋白融合成功并成功展示在细胞壁上。对比分析重组菌与野生菌的细胞内容物,发现重组菌一组中在108 kDa处(图 4中6泳道所示)有条带显示,这说明融合蛋白首先在胞内形成并很快展示到细胞壁上。并且对照重组菌发酵液上清和重组菌胞壁蛋白、重组菌细胞内容物,分析认为外源基因经α-factor信号肽准确表达引导[14],融合蛋白首先在细胞内的内质网形成,在高尔基体上修饰,然后向外传送使之锚定在细胞壁上,并且锚定牢固并未出现外源蛋白脱落的情况。

|
| 图 4 重组菌与野生菌发酵液上清、胞壁抽提蛋白以及胞内蛋白SDS-聚丙烯酰胺凝胶电泳对比 Fig. 4 Comparison of the supernatant of recombinant bacteria and wild mushroom fermentation liquid,cell wall extract protein and intracellular protein SDS- polyacrylamide gel electrophoresis 1: Recombinant bacteria fermentation supernatant protein determination; 2: Wild Mushroom fermentation supernatant protein determination; 3: Recombinant bacterial cell wall protein extraction determination; 4: Wild fungus cell wall protein extraction determination; 5: Recombinant bacteria cell contents protein determination; 6: Wild Mushroom cell contents protein determination; M:Protein ruler |
根据1.2.7 中的检测方法,利用 HPLC分别对重组菌发酵液上清,菌体沉淀重悬液,破碎后菌体内容物测得液相图谱结果如图 5所示。由HPLC测得液相图谱所示,图 5中A样品,为重组菌发酵液上清转化结果,未见麦芽糖转化为海藻糖,图 5中B样品,重组菌体沉淀重悬液和图 5中C样品,破碎后重组菌菌体内容物转化结果表明已有部分麦芽糖转化为海藻糖。而对照组转入空质粒的毕赤酵母D,E,F均未见海藻糖峰。这说明海藻糖合酶一部分在重组菌胞内表达;海藻糖合酶基因通过与Pir型锚定蛋白基因融合成功地在毕赤酵母表面展示,并具有一定酶活。由5图中A,B转化结果分析再次证明通过共价键结合在细胞壁上的Pir-C端锚定海藻糖合酶的结构很稳定,未见有融合蛋白分泌至发酵液上清中。由图 5中B,C转化结果进一步证实海藻糖合酶融合蛋白首先在胞内表达,然后锚定至酵母细胞壁上。

|
|
图 5 重组菌、野生菌发酵液上清,菌体沉淀重悬液, 破碎后菌体内容物转化生成海藻糖液相图谱
Fig. 5 HPLC results of intracellular and extracellular recombinants
Peak table from left to right as water peak,glucose peak,maltose peak,trehalose peak. A:Supernatant of recombinant bacterial fermentation broth; B:Recombinant bacterial precipitation of heavy suspension; C:The activity of the contents of the recombinant bacteria in vivo; D:Control bacteria fermentation broth supernatant enzyme activity; E:The activity of the precipitation of the wild strains by the precipitation of heavy suspension; F:The contents of the wild mushroom after the break |
以终浓度为30%的麦芽糖为底物,pH 7.5的条件下,于不同温度条件下测定表面展示海藻糖合酶酶活。如图 6所示最适反应温度为50℃,此时酶活能够达到300.65 U/mg,随着温度升高,酶活显著降低。表面展示海藻糖合酶的温度耐受性实验结果如图 7所示,40℃条件下,保温60 min,酶活基本没有损失,60 ℃保温60 min之后酶活下降幅度显著,这说明表面展示海藻糖合酶在40 ℃以内具有良好的耐热性。

|
| 图 6 温度对酶活的影响 Fig. 6 Effect of temperature on enzyme activity |

|
| 图 7 表面展示海藻糖合酶的温度耐受性 Fig. 7 Temperature tolerance of surface display trehalose synthase |
以终浓度为30%的麦芽糖为底物,50 ℃的条件下,于不同pH条件下测定表面展示海藻糖合酶酶活。如图 8所示最适反应pH为7.5,反应条件偏酸不利于酶促反应,该酶耐酸性较差。对表面展示海藻糖合酶的pH耐受性实验显示,pH 7.0,7.5,8.0三种条件下耐受60 min后相对酶活均保持在50%以上,其中pH 8.0的偏碱环境下耐受60 min后相对酶活保持在73%以上,这说明表面展示海藻糖合酶对碱性环境有很强的耐受性(图 9)。

|
| 图 8 pH对酶活的影响 Fig. 8 Effect of pH on enzyme activity |

|
| 图 9 表面展示海藻糖合酶的pH耐受性 Fig. 9 pH tolerance of surface display trehalose synthase |
实验成功构建了来源于Pseudomonas putide P06菌株的海藻糖合酶基因和来源于酿酒酵母的pir1p基因的毕赤酵母表面展示重组菌,首次利用Pir1p蛋白共价连接[14]于酵母细胞壁上的特征,将Tres与Pir1p蛋白融合并展示于毕赤酵母表面,融合蛋白在毕赤酵母GS115中得到初步表达。重组毕赤酵母可以直接利用菌体作为全细胞催化剂与麦芽糖反应,避免了破碎细胞提纯海藻糖合酶的繁琐步骤,节约生产成本,利于工业化生产。本实验通过SDS聚丙烯酰胺凝胶电泳的方法简单检测到了海藻糖合酶存在,通过HPLC方法再次证明海藻糖合酶在毕赤酵母中表达成功并锚定在酵母细胞壁上,且检测到一定的酶活。表面展示海藻糖合酶在最优反应条件50 ℃,pH 7.5条件下酶活达到300.65 U/g,偏酸环境不利于酶促反应进行。对该酶的温度耐受性和pH耐受性实验表明,该酶在40 ℃范围内保温60min酶活稳定,剩余相对酶活达75%以上;该酶具有较好的pH耐受性,在pH 7.5~8.0范围内处理60 min之后,剩余相对酶活保持在72%以上,但未来融合蛋白的产量及海藻糖合酶酶活的提高方面仍有较大的探索空间。
另外,毕赤酵母重组菌在中试试验中诱导产融合蛋白的最适温度、pH、及供氧量乃至最适发酵周期等,这些因素都将对发酵效率产生影响[15, 16, 17]。外源蛋白的成功表达涉及基因转录、翻译、蛋白质折叠加工、囊泡运输等分泌表达过程的多个环节,是一个精细调控的复杂系统[18, 19]。初生多肽链在内质网中的折叠、加工等是影响外源蛋白分泌表达的限速步骤[19, 20, 21],未来应对外源蛋白的分泌途径中加工修饰的分子机制进行深入探索。
| [1] | Elbein A D,Pan Y T,Pastuazak I,ei al. New insights on trehalose; a multifunctional molecule. Glycobiology,2003,13(4):17-27. |
| [2] | 段作营,姚林,毛忠贵.Pseudomonas putida S1海藻糖合成酶基因在大肠杆菌中的克隆表达.工业微生物,2008,38(6):7-12. Duan Z Y,Yao L,Mao Z G. Cloning and expression of trehalose synthase gene from Pseudomonas putida S1 in Escherichia coli. Industrial Microbiology,2008,38(6):7-12. |
| [3] | 郝昭程,王腾飞,李忠奎,等. 拟南芥硫酯酶基因在毕赤酵母中的表达. 生物工程学报,2015,31(2):1-8. Hao Z C,Wang T F,Li Z K,et al. Expression of Arabidopsis thaliana thioesterase gene in Pichia pastoris. Chin J Biotech,2015,31(2):1-8. |
| [4] | Charoenrat T,Ketudat-Cairns M,Stendahl-Andersen H,et al. Oxygen-limitedfed-batch process:an alternative control for Pichia pastoris recombinant protein processes. Bioprocess Biosyst Eng,2005,27(6):399-406. |
| [5] | 苟兴华,王卫,刘达玉,等. 麦芽寡糖基海藻糖水解酶基因在巴斯德酵母中的表达及遗传稳定性. 应用与环境生物学报,2010,16(3):408-411. Gou X H,Wang W,Liu D Y,et al. Expression of MTHase gene in Pichia pastoris and its genetic stability. Chinese Journal of Applied and Environmental Biology,2010,16(3):408-411. |
| [6] | 张卉,袁其朋.Hepcidin的基因克隆及其在毕赤酵母中的分泌表达.生物工程学报,2007,23(3):381-385. Zhang H,Yuan Q P. Cloning and secretion expression of hepcidin in Pichia pastoris. Chinese Journal of Biotechnology,2007,23(3):381-385. |
| [7] | 杨蕾蕾,袁其朋,李文进,等.玫瑰微球菌中treZ基因在毕赤酵母中的表达研究.生物技术通报,2009,10:173-177. Yang L L,Yuan Q P,Li W J,et al. Secretory expression of maltooligosyl trehalose trehalohydrolase Pichia pastoris from Micrococcus roseus QS412. Biotechnology Bulletin,2009,10:173-177. |
| [8] | 韩雪清,刘湘涛,尹双辉. 毕赤酵母表达系统. 微生物学杂志,2003,23(4):35-53. Han X Q,Liu X T,Yin S H. Expression system of Pichia pastoris. Journal of Microbiology,2003,23(4):35-53. |
| [9] | Maeauley-Patriek S,Fazenda M L,McNeil B,et al. Heterologous protein production using the Pichia pastoris expression system. Yeast,2015,22(4):249-270. |
| [10] | 刘俊梅,聂海彦,郑薇薇,等.水生栖热菌FL-03海藻糖合酶基因的克隆及真核表达.食品科学,2010,31(23):267-270. Liu J M,Nie H Y,Zheng W W,et al. Cloning and eukaryotic expression of trehalose synthase gene from Thermus aquaticus FL-03. Food Science,2010,31(23):267-270. |
| [11] | 陶站华,王凤芝. 酵母表面展示酶技术. 现代生物医学进展,2010,10(3):593-596. Tao Z H,Wang F Z. Yeast surface display technology. Progress in Modern Biomedicine,2010,10(3):593-596. |
| [12] | Gasser B,Sauer M,Maurer M,et al. Transcriptomics-based identification of novel factors enhancing heterologous protein secretion in yeasts. Applied and Environmental Microbiology,2007,73(20):6499-6507. |
| [13] | Daly R,Heam M T. Expression of heterologous proteins in Pichia pastoris:a useful experimenial tool in protein engineering and production. J Mol Reeognit,2005,18(2):119-138. |
| [14] | De Groot P W,Ram A F,Klis F M. Features and functions of covalently linked proteins in fungal cell walls. Fungal Genet Biol,2005,42(8):657-675. |
| [15] | Fernandez M L,Murga A.Influence of the incubation temperature on the autolytic activity of Lactobacillus acidophilus.Journal of Applied Bacteriology,1995,78:426-429. |
| [16] | Zhu T,You L,Gong F,et al. Combinatorial strategy of sorbitol feeding and low-temperature induction leads to high-level production of alkaline β-mannanase in Pichia pastoris. Enzyme Microb Technol,2011,49(4):407-412. |
| [17] | Jiang F,Kongsaeree P,Schilke K,et al. Effects of pH and temperature on recombinant manganese peroxidase production and stability. Appl Biochem Biotechnol,2008,146(1/3):15-27. |
| [18] | 关波,金坚,李华钟.改良毕赤酵母分泌表达外源蛋白能力的研究进展.微生物报,2011,51(7):851-857. Guan B,Jin J,Li H Z. Genetic engineering of Pichia pastoris expression system for improved secretion of heterologous proteins. Acta Microbiologica Sinica,2011,51(7):851-857. |
| [19] | Leonardo M Damasceno,Chung Jr Huang,Carl A Batt.Protein secretion in Pichia pastoris and advances in protein production. Appl Microbiol Biotechnol,2012,93(1):31-39. |
| [20] | Inan M,Aryasomayajula D,Sinha J,et al. Enhancement of protein secretion in Pichia pastoris by overexpression of protein disulfide isomerase. Biotechnology and Bioengineering,2006,93(4):771-778. |
| [21] | Xu P,Robinson A S. Decreased secretion and unfolded protein response up-regulation are correlated with intracellular retention for single-chain antibody variants produced in yeast. Biotechnology and Bioengineering,2009,104(1):20-29. |