 2023, Vol. 22
2023, Vol. 22



2) Marine Biomedical Research Institute of Qiangdao, Qingdao 266237, China;
3) Laboratory for Marine Drugs and Bioproducts, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266237, China
Janus Kinases (JAKs) are a key family of cytoplasmatic protein tyrosine kinases which play a central role in cytokine signal transduction or growth factor receptor activation. Up to now, JAK1, JAK2, JAK3 and tyrosine kinase 2 (TYK2), as for the closely related four members of JAK family of enzymes, have been confirmed in mammalian (Clark et al., 2014; Clere-Jehl et al., 2020). Binding of the cytokines to their corresponding receptors induces JAK activation, and subsequently the Signal Transducers and Activators of Transcription (STAT) enzymes with downstream recruitment are phosphorylated, leading to the dimerization of the STATs. The STAT dimers undergo translocation to the nucleus, and then regulate the expression of target genes. Since the JAK/STAT pathway plays a major role in many basic processes, such as apoptosis and inflammation, so the dysfunctional proteins in this pathway may lead to a variety of diseases, including cancer and diseases that affect the immune system, such as severe combined immune deficient (SCID) (Flanagan et al., 2010; Schwartz et al., 2016).
JAK1, JAK2, and TYK2 are ubiquitously expressed in various tissues and cells of vertebrates, and JAK3 is pre-dominantly expressed in bone marrow cells, thymocytes, NK cells, activated B lymphocytes and T lymphocytes. JAK3 drives pro-inflammatory signaling cascades, including cytokine expression in the synovial fibroblasts and activited monocytes and macrophage. JAK3 associates with type Ⅰ cytokine receptors involving in a common γ-chain (γc) subunit, such as receptors for interleukin 2 (IL2), IL4, IL7, IL9, IL15 and IL21, which is important in T lymphocyte development, proliferation and differentiation (Verbsky et al., 1996; Chen et al., 1997; Elliott et al., 2011). Based on the functional characteristics and special tissue distribution of each subtype of the JAK kinase family, JAK1 is regarded as a novel target in the fields of immunity, inflammation and cancer, while JAK2 is considered as an effective target for the treatment and prevention of hematology-related diseases, and JAK3 has become a popular target for the treatment of autoimmune diseases (O'Shea et al., 2013).
So far, many JAK inhibitors have been approved by the US Food and Drug Administration (FDA) and other agencies, or have been evaluated in clinical trials, such as To-facitinib (1, a first-in-class JAK3 inhibitor approved by FDA in 2012) (Changelian et al., 2003; West, 2009), Ruxolitinib (2, a JAK1/2 inhibitor approved by FDA in 2011) (Harrison et al., 2012), Peficitinib (3, a JAK3 inhibitor approved by PMDA in 2019) (Ito et al., 2017), Pacritinib (4, a JAK2/ FLT3 inhibitor, Phase Ⅲ) (Jensen et al., 2017), Decernotinib (5, a JAK3 inhibitor, Phase Ⅲ) (Fleischmann, 2012), Cerdulatinib (6, a JAK1/JAK3/TYK2 inhibitor) (Blunt et al., 2016; Coffey et al., 2019), CYT387 (7, a JAK1/2 inhibitor) (Monaghan et al., 2011), AZD1480 (8, a JAK1/2 inhibitor) (Lu et al., 2017), PF-06651600 (9, a JAK3 inhibitor, Phase Ⅲ) (Thorarensen et al., 2017), and others (Hart et al., 2011) (Fig. 1). JAK inhibitors for clinical use exhibit certain side effects due to pan inhibition. Therefore, current researches mainly focus on the development of highly selective JAK inhibitors. Recent studies have exhibited that the JAK3 protein is different from the other JAKs, the active site of JAK3 is unique as it contains a cysteine (Cys909) which if properly engaged could provide access to JAK3 selective inhibitors (Liu et al., 2013; Barua et al., 2017). Based on these reserches, many studies have shown that JAK3 inhibitors, which were designed against Cys909, exhibited higher selectivity than those inhibitors for other three JAKs (Thorarensen et al., 2017).

|
Fig. 1 Structures of some JAK inhibitors. |
Marine biological resources have become an important source of lead compounds in new drug discovery and development due to their diversity, complexity and speciality. Up to now, up to 17 clinical available marine drugs have been developed from marine natural products or their derivatives (Shinde et al., 2019). Neobacillamide A (10), a marine alkaloid isolated from the greedy and stubborn sponge symbiotic Bacillus atrophicus C89 in the South China Sea (Fig. 2) (Yu et al., 2009), was predicted that its binding energy is < −9 kcal mol against corresponding target of JAK3 by virtual screening in smart supercomputing platforms of the Pilot Ocean National Laboratory (MarinChem-3D, http://mc3d.qnlm.ac/), suggesting Neobacillamide A (10) may as the hit to exploit selectively JAK3 inhibitors. Recently, several researchers have disclosed their studies on novel JAK3 selective inhibitors. For example, Rajesh Bahekar et al. (2020) discovered a potent and orally bioavailable 2, 4-diaminepyrimidine-5-carboxamide based JAK3 selective inhibitor, and Yin et al. (2020) identified a series of pyrazolopyrimidine derivatives as potent JAK3 inhibitor, and so on. In this paper, considering the active groups of these highly selective JAK3 inhibitor, we designed and synthesized a series of Neobacillamide A derivatives with modification at 2- and 4-positions of thiazole ring, which were evaluated for their JAK/STAT inhibitory activities and anti-tumor activities to A549 cells.

|
Fig. 2 Structure of Neobacillamide A. |
Commercial reagents were used without further purification unless specialized. Thin-layer chromatography (TLC) was performed on precoated E. Merck Silica Gel 60 F254 plates. Flash column chromatography was performed on silica gel (200–300 mesh). Optical rotations were determined with a Perkin-Elmer Model 241 MC polarimeter. 1H NMR and 13C NMR spectra were taken on a JEOL JNM-ECP 600 spectrometer with tetramethylsilane as the internal standard, and chemical shifts are recorded in values. Mass spectra were recorded on a Q-TOF Global mass spectrometer.
2.2 Chemical Methods 2.2.1 General procedure for the synthesis of compounds 13a – 13o and 15To the solution of reagent 23 (180 mg, 0.84 mmol) in CH2Cl2 (15 mL), TEA (0.29 mL, 2.10 mmol) and EDCI (241.5 mg, 1.26 mmol) were added, and the reaction mixture was stirred for 30 min at room temperature. Then HOBt (113.5 mg, 0.84 mmol) and substituted amines (0.84 mmol) were added to the reaction solution under nitrogen atmosphere. The reaction mixture was stirred overnight at room temperature. After completion of reaction, the mixture was diluted with water and extracted with CH2Cl2. The organic layer was dried over MgSO4, filtered and concentrated under vacuum to afford the crude product, which was purified by flash chromatography (CH2Cl2: MeOH = 20:1) to provide the desired title products 13a – 13o and 15.
2.2.1.1 (S)-(N-(2-phenethyl))-2-(1-acetamidoethyl) thiazole-4-carboxamide (13a)Refer to general procedure as described in Section 2.2.1. 48.3% isolated yield, white solid; m.p. 153 – 154℃; 1H NMR (400 MHz, Chloroform-d) δ 8.02 (s, 1H), 7.34 (dd, J = 16.0, 8.0 Hz, 3H), 7.27 (s, 2H), 7.24 (d, J = 7.3 Hz, 1H), 6.04 (d, J = 6.9 Hz, 1H), 5.42 – 5.36 (m, 1H), 3.70 (q, J = 7.0 Hz, 2H), 2.94 (t, J = 7.1 Hz, 2H), 2.05 (s, 3H), 1.60 (d, J = 6.9 Hz, 3H); HRMS (ESI) m /z: calcd for (C16H20O2N3S + H)+, 318.1271; found: 318.1273.
2.2.1.2 (S)-(N-phenyl)-2-(1-acetamidoethyl)thiazole-4-carboxamide (13b)Refer to general procedure as described in Section 2.2.1. 80.1% isolated yield, white solid; m.p. 138 – 139℃; [α]D20 = −10.13 (c = 1.10, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 10.06 (s, 1H), 8.75 (d, J = 7.4 Hz, 1H), 8.31 (s, 1H), 7.81 (d, J = 7.5 Hz, 2H), 7.39 – 7.32 (m, 2H), 7.11 (t, J = 7.4 Hz, 1H), 5.25 – 5.18 (m, 1H), 1.92 (s, 3H), 1.56 (d, J = 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.7 (C), 169.3 (C), 159.2 (C), 149.5 (C), 138.4 (C), 128.7 (2CH), 124.8 (CH), 124.0 (CH), 120.6 (2CH), 47.0 (CH), 22.6 (CH3), 20.8 (CH3); HRMS (ESI) m /z: calcd for (C14H16O2N3S + H)+, 290.0958; found: 290.0965.
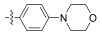
2.2.1.3 (S)-(N-morpholine phenyl)-2-(1-acetami-doethyl)thiazole-4-carboxamide (13c)Refer to general procedure as described in Section 2.2.1. 57.8% isolated yield, white solid; m.p. 204 – 205℃; [α]D20 = −10.13 (c = 1.10, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 9.89 (s, 1H), 8.73 (d, J = 7.4 Hz, 1H), 8.25 (s, 1H), 7.66 (d, J = 9.0 Hz, 2H), 6.93 (d, J = 9.0 Hz, 2H), 5.24 – 5.17 (m, 1H), 3.74 (t, 4H), 3.07 (t, 4H), 1.92 (s, 3H), 1.55 (d, J = 7.0 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 175.5 (C), 169.3 (C), 158.7 (C), 149.8 (C), 147.8 (C), 130.6 (C), 124.20 (CH), 121.6 (2CH), 115.31 (2CH), 66.2 (2CH2), 48.9 (2CH2), 47.0 (CH), 22.6 (CH3), 20.8 (CH3); HRMS (ESI) m /z: calcd for (C18H23O3N4S + H)+, 375.1485; found: 375.1481.
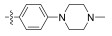
2.2.1.4 (S)-(N-methylpiperazine phenyl)-2-(1-aceta-midoethyl)thiazole-4-carboxamide (13d)Refer to general procedure as described in Section 2.2.1. 35.4% isolated yield, white solid; m.p. 175 – 176℃; [α]D20 = −3.21 (c = 0.72, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 9.87 (s, 1H), 8.73 (d, J = 7.4 Hz, 1H), 8.24 (s, 1H), 7.64 (d, J = 9.0 Hz, 2H), 6.92 (d, J = 9.0 Hz, 2H), 5.24 – 5.17 (m, 1H), 3.10 (t, 4H), 2.45 (t, 4H), 2.22 (s, 3H), 1.92 (s, 3H), 1.55 (d, J = 7.0 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.5 (C), 169.3 (C), 158.7 (C), 149.8 (C), 147.8 (C), 130.2 (C), 124.2 (CH), 121.6 (2CH), 115.5 (2CH), 54.7 (2CH2), 48.5 (2CH2), 47.0 (CH), 45.9 (CH3), 22.6 (CH3), 20.8 (CH3); HRMS (ESI) m /z: calcd for (C19H26O2N5S + H)+, 388.1802; found: 388.1793.
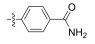
2.2.1.5 (S)-(N-(4-carbamoyl)-phenyl)-2-(1-acetami-doethyl)thiazole-4-carboxamide (13e)Refer to general procedure as described in Section 2.2.1. 38.6% isolated yield, white solid; m.p. 231 – 232℃; [α]D20 = −3.21 (c = 0.72, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 10.27 (s, 1H), 8.76 (d, 1H), 8.35 (s, 1H), 7.89 (q, J = 8.8 Hz, 5H), 7.27 (s, 1H), 5.25 – 5.18 (m, 1H), 1.93 (s, 3H), 1.56 (d, J = 7.0 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.8 (C), 169.3 (C), 167.4 (C), 159.3 (C), 149.3 (C), 141.1 (C), 129.5 (C), 128.3 (2CH), 125.3 (CH), 119.6 (2CH), 47.1 (CH), 22.6 (CH3), 20.8 (CH3); HRMS (ESI) m /z: calcd for (C15H17O3N4S + H)+, 333.1016; found: 333.1022.
2.2.1.6 (S)-(N-(4-methyl sulfonamide)-phenyl)-2-(1-acetamidoethyl)thiazole-4-carboxamide (13f)Refer to general procedure as described in Section 2.2.1. 38.7% isolated yield, white solid; m.p. 184 – 185℃; [α]D20 = −3.48 (c = 0.92, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 9.62 (s, 1H), 8.74 (d, J = 7.3 Hz, 1H), 8.29 (s, 1H), 7.77 (d, J = 8.7 Hz, 2H), 7.19 (d, J = 8.7 Hz, 2H), 5.25 – 5.16 (m, 1H), 2.96 (s, 3H), 1.92 (s, 3H), 1.55 (d, J = 9.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.8 (C), 169.5 (C), 159.2 (C), 149.6 (C), 135.0 (C), 134.2 (C), 124.9 (CH), 121.7 (2CH), 121.1 (2CH), 47.1 (CH), 22.6 (CH3), 20.9 (CH3); HRMS (ESI) m /z: calcd for (C15H19O4N4S2 + H)+, 383.0842; found: 383.0841.
2.2.1.7 (S)-(N-(3-methyl sulfonamide)-phenyl)-2-(1-acetamidoethyl)thiazole-4-carboxamide (13g)Refer to general procedure as described in Section 2.2.1. 35.2% isolated yield, white solid; m.p. 190 – 191℃; [α]D20 = −4.41 (c = 0.95, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 10.13 (s, 1H), 9.79 (s, 1H), 8.74 (d, J = 7.4 Hz, 1H), 8.32 (s, 1H), 7.75 (s, 1H), 7.54 (d, J = 8.1 Hz, 1H), 7.29 (t, J = 8.1 Hz, 1H), 6.94 (d, J = 8.0 Hz, 1H), 5.25 – 5.18 (m, 1H), 3.03 (s, 3H), 1.92 (s, 3H), 1.55 (d, J = 8.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.7 (C), 169.3 (C), 159.2 (C), 149.5 (C), 139.2 (C), 138.8 (C), 129.4 (CH), 125.0 (CH), 116.1 (CH), 111.8 (CH), 99.6 (CH), 47.1 (CH), 22.6 (CH3), 20.8 (CH3); HRMS (ESI) m /z: calcd for (C15H19O4N4S2 + H)+, 383.0842; found: 383.0843.
2.2.1.8 (S)-(N-(3-aminosulfonyl)-phenyl)-2-(1-ace-tamidoethyl)thiazole-4-carboxamide (13h)Refer to general procedure as described in Section 2.2.1. 28.9% isolated yield, white solid; m.p. 161 – 162℃; [α]D20 = −4.41 (c = 0.95, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 10.42 (s, 1H), 8.75 (d, J = 7.3 Hz, 1H), 8.43 (s, 1H), 8.35 (s, 1H), 7.98 (d, J = 7.4 Hz, 1H), 7.55 (q, J = 8.2 Hz, 2H), 7.38 (s, 2H), 5.24 – 5.20 (m, 1H), 1.93 (s, 3H), 1.56 (d, J = 6.9 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.8 (C), 169.3 (C), 159.5 (C), 149.2 (C), 144.6 (C), 138.9 (C), 129.4 (CH), 125.4 (CH), 123.6 (CH), 121.1 (CH), 117.6 (CH), 47.1 (CH), 22.6 (CH3), 20.7 (CH3); HRMS (ESI) m /z: calcd for (C14H17O4N4S2 + H)+, 369.0686; found: 369.0694.
2.2.1.9 (S)-(N-(2-fluorobenzyl))-2-(1-acetamidoethyl) thiazole-4-carboxamide (13i)Refer to general procedure as described in Section 2.2.1. 36.2% isolated yield, white solid; m.p. 161 – 162℃; [α]D20 = −4.41 (c = 0.95, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 8.82 (t, 1H), 8.71 (d, J = 7.3 Hz, 1H), 8.16 (s, 1H), 7.32 (q, J = 8.3 Hz, 2H), 7.16 (t, J = 9.7 Hz, 2H), 5.20 – 5.12 (m, 1H), 4.51 (d, 2H), 1.90 (s, 3H), 1.51 (d, J = 8.0 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.4 (C), 169.3 (C), 160.8 (C), 149.4 (C), 129.5 (C), 128.8 (C), 126.1 (CH), 124.4 (CH), 123.9 (CH), 115.2 (CH), 115.0 (CH), 46.9 (CH), 36.1 (CH2), 22.6 (CH3), 20.6 (CH3); HRMS (ESI) m /z: calcd for (C15H17O2N3FS + H)+, 322.1020; found: 322.1025.
2.2.1.10 (S)-(N-(3-fluorobenzyl))-2-(1-acetamido-ethyl)thiazole-4-carboxamide (13j)Refer to general procedure as described in Section 2.2.1. 31.8% isolated yield, white solid; m.p. 161 – 162℃; [α]D20 = −4.41 (c = 0.95, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 8.94 (t, 1H), 8.71 (d, J = 8.5 Hz, 1H), 8.16 (s, 1H), 7.35 (dq, J = 14.4, 7.9, 7.0 Hz, 1H), 7.18 – 7.01 (m, 3H), 5.21 – 5.11 (m, 1H), 4.46 (d, 2H), 1.90 (s, 3H), 1.51 (d, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.4 (C), 169.3 (C), 160.8 (C), 149.5 (C), 142.8 (C), 130.4 (C), 123.9 (CH), 123.5 (CH), 114.2 (CH), 114.0 (CH), 113.5 (CH), 46.9 (CH), 41.9 (CH2), 22.6 (CH3), 20.6 (CH3); HRMS (ESI) m /z: calcd for (C15H17O2N3FS + H)+, 322.1020; found: 322.1027.
2.2.1.11 (S)-(N-(3-benzothiazolyl))-2-(1-acetamido-ethyl)thiazole-4-carboxamide (13k)Refer to general procedure as described in Section 2.2.1. 32.2% isolated yield, white solid; m.p. 172 – 173℃; [α]D20 = −2.71 (c = 0.63, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 10.37 (s, 1H), 9.31 (s, 1H), 8.76 (d, J = 7.4 Hz, 1H), 8.70 (d, J = 1.9 Hz, 1H), 8.36 (s, 1H), 8.06 (d, J = 8.8 Hz, 1H), 7.92 (q, 1H), 5.27 – 5.20 (m, 1H), 1.93 (s, 3H), 1.57 (d, J = 7.0 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.8 (C), 169.3 (C), 159.4 (C), 155.2 (CH), 149.7 (C), 149.4 (C), 136.2 (C), 134.1 (C), 125.1 (CH), 122.9 (CH), 120.2 (CH), 113.3 (CH), 47.1 (CH), 22.6 (CH3), 20.8 (CH3); HRMS (ESI) m /z: calcd for (C15H15O2N4S2 + H)+, 347.0631; found: 347.0634.
2.2.1.12 (S)-(N-(5, 6, 7, 8-tetrahydropyrido[4, 3-d]pyri-midinyl))-2-(1-acetamidoethyl)thiazole-4-carboxamide (13l)Refer to general procedure as described in Section 2.2.1. 53.8% isolated yield, white solid; m.p. 206 – 207℃; [α]D20 = −2.71 (c = 0.63, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 8.68 (d, J = 7.8 Hz, 1H), 8.10 (t, J = 38.2 Hz, 2H), 6.49 (s, 2H), 5.21 – 5.17 (m, 1H), 4.66 (d, J = 56.0 Hz, 2H), 3.35 (t, 1H), 2.73 (s, J = 14.7 Hz, 2H), 1.90 (s, 3H), 1.50 (d, J = 9.0 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 174.0 (C), 169.1 (C), 163.4 (C), 162.3 (CH), 156.2 (C), 149.2 (C), 124.5 (C), 124.1 (CH), 114.9 (C), 46.6 (CH), 43.7 (CH2), 41.3 (CH2), 31.8 (CH2), 22.6 (CH3), 20.3 (CH3); HRMS (ESI) m /z: calcd for (C15H19O2N6S + H)+, 347.1285; found: 347.1280.
2.2.1.13 (S)-(N-(1-Methyl-1H-pyrazol-3-yl))-2-(1-ace-tamidoethyl)thiazole-4-carboxamide (13m)Refer to general procedure as described in Section 2.2.1. 68.3% isolated yield, white solid; m.p. 170 – 172℃; [α]D20 = −7.75 (c = 1.17, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 10.34 (s, 1H), 8.73 (d, J = 7.2 Hz, 1H), 8.21 (s, 1H), 8.02 (s, 1H), 7.66 (s, 1H), 5.23 – 5.14 (m, 1H), 3.81 (s, 3H), 1.91 (s, 3H), 1.54 (d, J = 8.9 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.8 (C), 169.3 (C), 157.7 (C), 149.4 (C), 130.7 (C), 124.1 (CH), 121.9 (CH), 121.3 (CH), 47.1 (CH), 38.8 (CH3), 22.6 (CH3), 20.8 (CH3); HRMS (ESI) m /z: calcd for (C12H16O2N5S + H)+, 294.1019; found: 294.1024.
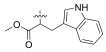
2.2.1.14 (S)-(N-(L-trptophan methyl ester))-2-(1-ace-tamidoethyl)thiazole-4-carboxamide (13n)Refer to general procedure as described in Section 2.2.1. 40.3% isolated yield, white solid; m.p. 84 – 85℃; [α]D20 = −7.75 (c = 1.17, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 10.92 (s, 1H), 8.70 (t, J = 6.9 Hz, 1H), 8.24 (d, J = 16.8, 7.7 Hz, 1H), 8.15 (d, J = 5.2 Hz, 1H), 7.51 (t, J = 6.9 Hz, 1H), 7.33 (d, J = 8.1 Hz, 1H), 7.17 (s, 1H), 7.06 (t, J = 7.7 Hz, 1H), 6.96 (q, J = 7.2 Hz, 1H), 5.16 – 5.09 (m, 1H), 4.76 (q, J = 6.4 Hz, 1H), 3.65 (s, 3H), 3.30 (s, 1H), 1.89 (d, J = 3.5 Hz, 3H), 1.47 (d, J = 7.0, 3.4 Hz, 3H), 1.23 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.0 (C), 169.3 (C), 160.3 (C), 148.7 (C), 136.2 (C), 127.2 (C), 124.3 (C), 123.8 (CH), 121.2 (CH), 118.6 (CH), 118.2 (CH), 111.6 (CH), 109.4 (CH), 109.2 (C), 52.2 (CH), 46.9 (CH3), 26.8 (CH), 22.6 (CH2), 20.5 (CH3), 20.3 (CH3); HRMS (ESI) m /z: calcd for (C20H23O4N4S + H)+, 415.1435; found: 415.1434.
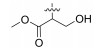
2.2.1.15 (S)-(N-(L-serine methyl ester))-2-(1-aceta-midoethyl)thiazole-4-carboxamide (13o)Refer to general procedure as described in Section 2.2.1. 59.1% isolated yield, white solid; m.p. 154 – 155℃; [α]D20 = −7.75 (c = 1.17, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 8.75 (d, J = 7.1 Hz, 1H), 8.21 (s, 1H), 8.18 (d, J = 6.6 Hz, 1H), 5.29 (q, 1H), 5.21 – 5.12 (m, 1H), 4.60 – 4.53 (m, 1H), 3.87 (t, J = 7.7 Hz, 1H), 3.76 (d, J = 12.5 Hz, 1H), 3.67 (s, 3H), 1.90 (s, 3H), 1.51 (d, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.7 (C), 170.9 (C), 169.3 (C), 160.3 (C), 148.8 (C), 124.4 (CH), 61.2 (CH2), 54.6 (CH), 52.2 (CH3), 46.9 (CH), 22.6 (CH3), 20.6 (CH3); HRMS (ESI) m /z: calcd for (C12H18O5N3S + H)+, 316.0962; found: 316.0969.
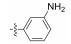
2.2.2 (S)-(N-(3-aminophenyl))-2-(1-acetamidoethyl) thiazole-4-carboxamide (13p)To a solution of 15 (5.40 g, 13.4 mmol) in CH2Cl2 (25 mL) was added trifluoroacetic acid (10 mL) at 0℃, and the reaction mixture was stirred for 30 min at room temperature. Then, TFA was evaporated under vacuum. The recry-stallization of the crude product was carried out by using MeOH, to give 13p (3.70 g, 91.1%) as a white solid; m.p. 162 – 163℃; [α]D20 = −11.42 (c = 0.74, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 9.66 (s, 1H), 8.73 (d, J = 7.5 Hz, 1H), 8.26 (s, 1H), 7.10 (s, 1H), 6.97 (t, J = 7.9 Hz, 1H), 6.86 (d, J = 8.0 Hz, 1H), 6.33 (d, J = 9.1 Hz, 1H), 5.24 – 5.11 (m, 1H), 5.11 (s, 2H), 1.92 (s, 3H), 1.55 (d, J = 7.0 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.3 (C), 169.3 (C), 158.8 (C), 149.7 (C), 149.1 (C), 138.9 (C), 129.0 (CH), 124.4 (CH), 110.1 (CH), 108.3 (CH), 99.6 (CH), 47.0 (CH), 22.6 (CH3), 20.7 (CH3); HRMS (ESI) m /z: calcd for (C14H17O2N4S + H)+, 305.1067; found: 305.1061.
2.2.3 General procedure for the synthesis of compounds 14a – 14e and 14hDIPEA (0.11 mL, 0.66 mmol) and HATU (627 mg, 1.65 mmol) were added to the solution of 13p (200 mg, 0.66 mmol) in DMF (15 mL), then different substituted acids (0.79 mmol) were added to the reaction solution under N2. The reaction mixture was stirred overnight at room temperature. After completion of reaction, the mixture was diluted with water and extracted with CH2Cl2. The organic layer was dried over MgSO4, filtered and concentrated under vacuum to afford the crude product, which was purified by flash chromatography (CH2Cl2: MeOH = 30:1) to provide the desired title products 14a – 14e and 14h.
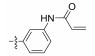
2.2.3.1 (S)-(N-(3-acrylamidyl))-2-(1-acetamidoethyl) thiazole-4-carboxamide (14a)Refer to general procedure as described in Section 2.2.3. 47.2% isolated yield, white solid; m.p. 207 – 208℃; [α]D20 = −11.42 (c = 0.74, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 10.19 (s, 1H), 10.05 (s, 1H), 8.74 (d, 1H), 8.31 (s, 1H), 8.18 (s, 1H), 7.47 (dd, J = 12.7, 8.8 Hz, 2H), 7.29 (t, J = 8.1 Hz, 1H), 6.47 (dd, J = 17.0, 10.1 Hz, 1H), 6.26 (dd, J = 17.0, 1.9 Hz, 1H), 5.75 (dd, J = 10.1, 1.8 Hz, 1H), 5.25 – 5.18 (m, 1H), 1.92 (s, 3H), 1.56 (d, J = 7.0 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.5 (C), 169.3 (C), 163.2 (C), 159.1 (C), 149.5 (C), 139.3 (C), 138.7 (C), 132.0 (CH), 129.0 (CH), 126.9 (CH2), 124.8 (CH), 115.9 (CH), 115.3 (CH), 111.7 (CH), 47.0 (CH), 22.6 (CH3), 20.7 (CH3); HRMS (ESI) m /z: calcd for (C17H19O3N4S + H)+, 359.1172; found: 359.1171.
2.2.3.2 (S)-(N-(3-propionamidyl))-2-(1-acetamido-ethyl)thiazole-4-carboxamide (14b)Refer to general procedure as described in Section 2.2.3. 46.3% isolated yield, white solid; m.p. 202 – 203℃; [α]D20 = −11.42 (c = 0.74, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 10.00 (s, 1H), 9.90 (s, 1H), 8.74 (d, J = 7.4 Hz, 1H), 8.30 (s, 1H), 8.10 (s, 1H), 7.45 – 7.34 (m, 2H), 7.25 (t, J = 8.1 Hz, 1H), 5.23 – 5.18 (m, J = 7.1 Hz, 1H), 2.32 (q, J = 7.9 Hz, 2H), 1.92 (s, 3H), 1.56 (d, J = 6.9 Hz, 3H), 1.08 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.5 (C), 172.1 (C), 169.3 (C), 159.1 (C), 149.5 (C), 139.7 (C), 138.6 (C), 128.8 (CH), 124.8 (CH), 115.4 (CH), 111.5 (CH), 99.6 (CH), 47.0 (CH), 29.6 (CH2), 22.6 (CH3), 20.7 (CH3), 9.8 (CH3); HRMS (ESI) m /z: calcd for (C17H21O3N4S + H)+, 361.1329; found: 361.1332.
2.2.3.3 (S)-(N-(3-crotonic acid amidyl))-2-(1-aceta-midoethyl)thiazole-4-carboxamide (14c)Refer to general procedure as described in Section 2.2.3. 51.9% isolated yield, white solid; m.p. 208 – 209℃; [α]D20 = −11.42 (c = 0.74, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 10.02 (s, 1H), 9.99 (s, 1H), 8.74 (d, J = 7.5 Hz, 1H), 8.31 (s, 1H), 8.14 (s, 1H), 7.44 (t, J = 8.0 Hz, 2H), 7.26 (t, J = 8.1 Hz, 1H), 6.83 – 6.75 (m, 1H), 6.15 (d, J = 15.2 Hz, 1H), 5.23 – 5.20 (m, J = 7.2 Hz, 1H), 1.92 (s, 3H), 1.87 (d, J = 6.8 Hz, 3H), 1.56 (d, J = 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.5 (C), 169.3 (C), 163.6 (C), 159.1 (C), 149.5 (C), 139.9 (CH), 139.6 (C), 138.6 (C), 128.9 (CH), 126.1 (CH), 124.8 (CH), 115.7 (CH), 115.2 (CH), 111.7 (CH), 47.0 (CH), 22.6 (CH3), 20.7 (CH3), 17.6 (CH3); HRMS (ESI) m /z: calcd for (C18H21O3N4S + H)+, 373.1329; found: 373.1339.
2.2.3.4 (S)-(N-(3-methacrylic acid amidyl))-2-(1-ace-tamidoethyl)thiazole-4-carboxamide (14d)Refer to general procedure as described in Section 2.2.3. 48.5% isolated yield, white solid; m.p. 224 – 225℃; [α]D20 = −11.42 (c = 0.74, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 10.01 (s, 1H), 9.83 (s, 1H), 8.74 (d, J = 8.0 Hz, 1H), 8.31 (s, 1H), 8.18 (s, 1H), 7.48 (d, J = 8.3 Hz, 1H), 7.42 (d, J = 8.1 Hz, 1H), 7.28 (t, J = 8.0 Hz, 1H), 5.82 (s, 1H), 5.51 (s, 1H), 5.23 – 5.18 (m, 1H), 1.96 (s, 3H), 1.92 (s, 3H), 1.56 (d, J = 8.8 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.5 (C), 169.3 (C), 166.9 (C), 159.0 (C), 149.5 (C), 140.4 (C), 139.3 (C), 138.5 (C), 128.7 (CH), 124.8 (CH), 120.1 (CH2), 116.2 (CH), 116.0 (CH), 112.9 (CH), 47.0 (CH), 22.6 (CH3), 20.7 (CH3), 18.9 (CH3); HRMS (ESI) m /z: calcd for (C18H21O3N4S + H)+, 373.1329; found: 373.1332.
2.2.3.5 (S)-(N-(3-cyanoacetic amidyl))-2-(1-acetami-doethyl)thiazole-4-carboxamide (14e)Refer to general procedure as described in Section 2.2.3. 54.8% isolated yield, white solid; m.p. 237 – 238℃; [α]D20 = −11.42 (c = 0.74, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H), 10.10 (s, 1H), 8.75 (d, J = 8.2 Hz, 1H), 8.31 (s, 1H), 8.10 (s, 1H), 7.46 (d, J = 8.0 Hz, 1H), 7.36 (d, J = 8.1 Hz, 1H), 7.30 (t, J = 8.5 Hz, 1H), 5.24 – 5.19 (m, 1H), 3.90 (s, 2H), 1.92 (s, 3H), 1.56 (d, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.6 (C), 169.4 (C), 161.1 (C), 159.2 (C), 149.5 (C), 138.9 (C), 138.7 (C), 129.1 (CH), 124.9 (C), 116.3 (CH), 116.1 (CH), 115.0 (CH), 111.6 (CH), 47.0 (CH), 26.8 (CH2), 22.6 (CH3), 20.8 (CH3); HRMS (ESI) m /z: calcd for (C17H18O3N5S + H)+, 372.1125; found: 372.1127.
2.2.3.6 (S)-(N-(3-trans-cinnamic acid amidyl))-2-(1-acetamidoethyl)thiazole-4-carboxamide (14h)Refer to general procedure as described in Section 2.2.3. 43.5% isolated yield, white solid; m.p. 233 – 234℃; [α]D20 = −11.42 (c = 0.74, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 10.27 (s, 1H), 10.07 (s, 1H), 8.75 (d, J = 7.4 Hz, 1H), 8.32 (s, 1H), 8.20 (s, 1H), 7.59 (dd, J = 31.0, 9.2 Hz, 4H), 7.44 (t, J = 9.5 Hz, 4H), 7.31 (t, J = 8.1 Hz, 1H), 6.88 (d, J = 15.7 Hz, 1H), 5.26 – 5.19 (m, 1H), 1.93 (s, 3H), 1.57 (d, J = 7.6 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.5 (C), 169.3 (C), 163.6 (C), 159.1 (C), 149.5 (C), 140.2 (CH), 139.6 (C), 138.7 (C), 134.8 (C), 129.9 (CH), 129.1 (2CH), 129.0 (CH), 127.8 (2CH), 124.8 (CH), 122.4 (CH), 115.9 (CH), 115.1 (CH), 111.6 (CH), 47.0 (CH), 22.6 (CH3), 20.7 (CH3); HRMS (ESI) m /z: calcd for (C23H23O3N4S + H)+, 435.1485; found: 435.1488.
2.2.4 (S)-(N-(3-trifluoroacetyl))-2-(1-acetamidoethyl) thiazole-4-carboxamide (14f)To a solution of 13p (200 mg, 0.66 mmol) in THF (20 mL) was added TEA (0.14 mL, 0.99 mmol) and trifluoro-acetic anhydride (153 mg, 0.73 mmol) at 0℃, and the reaction mixture was stirred for overnight at room temperature. To the resulting solution was added aq NaHCO3 solution. The organic layer was dried over MgSO4 and evaporated under vacuum. The residue was purified by silica gel column chromatography (CH2Cl2: MeOH = 50:1), to give 14f (184 mg, 69.5%) as a white solid; m.p. 221 – 222℃; [α]D20 = −1.52 (c = 0.86, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 11.32 (s, 1H), 10.19 (s, 1H), 8.75 (d, J = 7.3 Hz, 1H), 8.58 – 8.20 (m, 2H), 7.61 (d, J = 7.6 Hz, 1H), 7.43 – 7.36 (m, 2H), 5.25 – 5.20 (m, 1H), 1.93 (s, 3H), 1.56 (d, J = 7.4 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.7 (C), 169.3 (C), 159.3 (C), 149.4 (C), 139.0 (C), 136.6 (C), 129.2 (CH), 125.1 (CH), 118.0 (CH), 116.9 (CH), 113.5 (CH), 99.6 (C), 47.0 (CH), 22.6 (CH3), 20.7 (CH3); HRMS (ESI) m /z: calcd for (C16H16O3N4F3S + H)+, 401.0890; found: 401.0884.
2.2.5 (S)-(N-(3-methyl oxalamidyl))-2-(1-acetamido-ethyl)thiazole-4-carboxamide (14g)To a solution of 13p (200 mg, 0.66 mmol) in CH2Cl2 (20 mL) was added TEA (0.01 mL, 0.66 mmol) and monome-thyl oxalate (0.07 mL, 0.73 mmol) at 0℃, and the reaction mixture was stirred for 30 min at room temperature. To the resulting solution was added MeOH. The mixture was concentrated under reduced pressure, and then the residue was purified by silica gel column chromatography (CH2Cl2: MeOH = 40:1), to give 14g (81 mg, 31.4%) as a white solid; m.p. 251 – 252℃; [α]D20 = −1.52 (c = 0.86, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 10.83 (s, 1H), 10.11 (s, 1H), 8.74 (d, J = 7.4 Hz, 1H), 8.32 (s, 1H), 8.26 (s, 1H), 7.56 (d, J = 8.1 Hz, 1H), 7.45 (d, J = 8.1 Hz, 1H), 7.33 (t, J = 8.1 Hz, 1H), 5.28 – 5.16 (m, 1H), 3.86 (s, 3H), 1.92 (s, 3H), 1.56 (d, J = 7.7 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.6 (C), 169.3 (C), 161.2 (C), 159.2 (C), 155.6 (C), 149.5 (C), 138.7 (C), 137.7 (C), 129.0 (CH), 124.9 (CH), 117.3 (CH), 116.5 (CH), 113.1 (CH), 53.2 (CH), 47.0 (CH3), 22.6 (CH3), 20.7 (CH3); HRMS (ESI) m/z: calcd for (C17H19O5N4S + H)+, 391.1071; found: 391.1069.
2.2.6 (S)-(N-(3-cinnamamidyl))-2-(1-acetamidoethyl) thiazole-4-carboxamide (14i)(Boc) 2O (2.38 g, 10.9 mmol) was added to the solution of 3-(aminomethyl)aniline 14i-1 (4.00 g, 32.7 mmol) in THF (100 mL), then the reaction mixture was stirred for overnight at room temperature. The reaction mixture was concentrated under reduced pressure, and then the residue was purified by silica gel column chromatography (EtOAc: PE = 4:1), to give 14i-2 (1.71 g, 70.4%) as a white solid.
TEA (2.33 mL, 16.9 mmol) and EDCI (1.94 g, 10.1 mmol) were added to the solution of trans-cinnamic acid (1.00 g, 6.75 mmol) in CH2Cl2 (30 mL) at 0℃. After sirred for 30 min, HOBt (912 mg, 6.75 mmol) and 14i-2 (825 mg, 6.75 mmol) were added to the reaction mixture under N2. The mixture was stirred for overnight. After completion of reaction, the mixture was diluted with water and extracted with CH2Cl2. The organic layer was dried over MgSO4, filtered and concentrated under vacuum to afford the crude product, which was purified by flash chromatography (EtOAc: PE = 1:3), to give 14i-3 (1.36 g, 57.2%) as a white solid; MS (ESI) m /z: calcd for (C21H24O3N2 + Na)+, 375.17; found: 375.02.
Trifluoroacetic acid (5 mL) was added to the solution of 14i-3 (1.20 g, 3.41 mmol) in CH2Cl2 (15 mL) at 0℃. The reaction was stirred for 30 min. Then, the mixture was concentrated under vacuum to afford the crude product 14i-4 (0.86 g, 89.8%) as a white solid. TEA (0.29 mL, 2.10 mmol) and EDCI (242 mg, 1.26 mmol) were added to a solution of 23 (180 mg, 0.84 mmol) in CH2Cl2 (15 mL) at 0℃. After stirred for 30 min, HOBt (114 mg, 0.84 mmol) and 14i-4 (212 mg, 0.84 mmol) were added to the reaction mixture under N2. The reaction was stirred for overnight. Then, the mixture was diluted with water and extracted with CH2Cl2. The organic layer was dried over MgSO4, filtered and concentrated under vacuum to afford the crude product, which was purified by flash chromatography (CH2Cl2: MeOH = 30:1), to give 14i (222 mg, 58.9%) as a white solid; m.p. 193 – 194℃; [α]D20 = −1.52 (c = 0.86, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, 1H), 8.87 (t, 1H), 8.71 (d, J = 7.5 Hz, 1H), 8.17 (s, 1H), 7.70 (d, J = 8.2 Hz, 1H), 7.65 – 7.50 (m, 4H), 7.43 (q, J = 8.1 Hz, 3H), 7.28 (t, J = 7.9 Hz, 1H), 7.02 (d, J = 7.6 Hz, 1H), 6.83 (d, J = 15.7 Hz, 1H), 5.20 – 5.15 (m, 1H), 4.45 (d, J = 6.0 Hz, 2H), 1.90 (s, 3H), 1.52 (d, J = 7.6 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 175.3 (C), 169.2 (C), 163.6 (C), 160.6 (C), 149.6 (C), 140.4 (C), 140.1 (CH), 139.4 (CH), 134.8 (CH), 129.8 (CH), 129.1 (2CH), 128.8 (CH), 127.8 (2CH), 123.7 (CH), 122.4 (CH), 118.0 (CH), 117.8 (CH), 99.6 (CH), 46.9 (CH), 42.3 (CH2), 22.6 (CH3), 20.6 (CH3); HRMS (ESI) m /z: calcd for (C24H25O3N4S + H)+, 449.1642; found: 449.1647.
2.3 Biological Analyses 2.3.1 Assay for cytotoxic activities against A549 cellsThe effect of the synthesized compound on the proliferation of A549 cells (BFN608007142, purchased from Solarbio, China) was eveluated by MTT assay. A549 cells in logarithmic growth period were selected when the confluence reaches 90%. A 96-well plate was seeded with 2000 cells per well in 100 μL of complete cell culture medium. After 24 h, 100 μL of complete medium containing 25 μmol L−1 of each compound was added to each well. Then, the cells were cultured for 72 h, followed by addition of 20 μL of resazurin (2 mg mL−1 dissolved in water) to the media and kept the rection for 8 h. The fluorescent signal was monitored at an excitation wavelength of 544 nm and an emission wavelength of 595 nm using a Spectramax M5 plate reader (Molecular Devices). The relative fluorescence unit (RFU) generated from the assay was proportional to the number of living cells in each well.
2.3.2 Molecular simulationDocking calculations were performed with Schrodinger program (version 11.8). The ligand and receptor preparation was performed under the ligand preparation and protein preparation module of Maestro 11.8. Then generating a Glid file to limit the docking position of ligands. The final step was molecular docking, and the flexible docking was performed and RMSD values were calculated.
3 Results and Discussion 3.1 Design of Neobacillamide A DerivativesIn the report of Bhide et al. (2017), compound 11 showed strong JAK3 inhibitory activity, with the IC50 value of 2 nmol L−1. The crystal structure (PDB code 5W86) of compound 11 and JAK3 showed that the hydrogen on the 5-position amide of the pyridine ring forms hydrogen bond with the amino acid residue, which may be the reason for the better activity (Fig. 3). Similarly, Bahekar et al. (2020) also retained the amide group at the 5-position of the pyrimidine ring and carried out structural modification at the 2-position, and finally obtained the most potent JAK3 inhibitor compound 12. According to the above studies, the amide side chain of C-2 of thiazole core is retained, and we modified the C-4 position with the active group of reference compounds, which showed superior JAK3 inhibitory activity (Bahekar et al., 2020; Yin et al., 2020), and finally a batch of Neobacillamide A derivatives were designed and scored by using Schrodinger software for molecular docking. Totally 24 novel derivatives with docking scores between −7.990 and −4.486 were filtered out (13a – 13o, 14a – 14i) and were chemically synthesized.

|
Fig. 3 Structure of compounds 11, 12 and crystal structure (PDB code 5W86) of compound 11 and JAK3. |
The synthesis of the designed Neobacillamide A derivatives 13a – 13p is shown in Scheme 1. According to the stereospecific strategy on synthesis of Neobacillamide A (Martínez and Danilo, 2013), the commercially available material Boc-L-(-)-alanine was reacted with di-tert-butyl dicarbonate (DIBOC) and ammonium bicarbonate to produce compound 17, which was then converted into nitrile 18 by dehydration with trifluoroacetic anhydride and pyridine. The condensation of 18 with L-cysteine ethylester hydrochloride under phosphate buffered conditions in methanol provide the thizoline 19, which was then oxidized to thiazole 20 by 1, 8-diazabicyclo[5.4.0]undec-7-ene (DBU) and bromotrichlo-romethane. The Boc group of 20 was hydrolyzed with tri-fluoroacetic acid to produce 21, followed by acetylated with acetic anhydride in pyridine providing 22, which was hy-drolyzed with 10% KOH solution to obtain intermediate 23. Finally, the condensation of 23 with substituted amines under EDCI and HOBt condition to furnish Neobacillamide A derivatives 13a – 13o and 15. The Boc group of 15 was hydrolyzed with trifluoroacetic acid to give Neobacillamide A derivative 13p.

|
Scheme1 Synthesis of compounds 13a – 13p. Reagents and conditions: (a) NH4HCO3, pyridine, (Boc)2O, 1, 4-dioxane, r.t.; (b) TFAA, pyridine, THF, 0℃ to r.t.; (c) L-cysteine ethyl hydrochloride, phosphate buffer pH 7, MeOH, reflux; (d) DBU, BrCCl3, CH2Cl2, −20℃ to −10℃; (e) TFA, CH2Cl2, r.t.; (f) Ac2O, pyridine, r.t.; (g) 10% aqueous KOH, THF, r.t.; (h) R1-NH2, EDCI/HOBt, TEA, CH2Cl2, r.t.; (i) TFA, CH2Cl2, r.t.. |
Another series of Neobacillamide A derivatives 14a – 14i were synthesized as depicted in Scheme 2. Condensation of 13p with substituted acids under HATU and DIPEA condition in DMF provided Neobacillamide A derivatives 14a – 14e and 14h. The derivatives 14f and 14g were obtained by reacting 13p with trifluoroacetic anhydride or methyl oxalyl chloride. The commercially available material 3-ami-nobenzylamine was acylated by (Boc)2O in THF, followed by condensation with trans-cinnamic acid using EDCI and HOBt, obtaining 14i-3, which was removed the Boc group with TFA producing intermediate 14i-4. Next, Coupling the intermediate 14i-4 with 23 provided Neobacillamide A derivatives 14i.

|
Scheme2 Synthesis of compounds 14a – 14h. Reagents and conditions: (a) R2COOH, DMF, DIPEA, HATU, 4 – 8 h, r.t.; (b) CF3OCOCOCF3, TEA, THF, 12 h, 0℃, r.t.; (c) CH3OCOCOCl, THF, TEA, 0℃, 30 min, r.t.; (d) THF, (Boc)2O, r.t., 8 h; (e) TEA, EDCI, HOBt, trans-cinnamic acid, DCM, r.t., 8 h; (f) TFA, DCM, 30 min; (g) TEA, EDCI, HOBt, DCM, r.t., 8 h. |
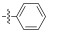
All synthesized Neobacillamide A derivatives were evaluated for their inhibitory activity against the JAK/STAT signaling pathway. Compounds 13c, 13o, 14d, 14g and 14h (concentration of 25 μmol L−1, all inhibitory potency were above 60%) showed potent inhibitory activity, especially, compound 14g exhibited the strongest activity, with the inhibition ratio of 89.70%. However, compounds 13b, 13d – 13n, 13p, 14a – 14c, 14e – 14f and 14i – 14j showed weak or no inhibitory activities against the JAK/STAT signaling pathway. From simple analysis, we found that the introduction of a 4-(4-morpholino) aniline, and L-serine methyl ester group into Neobacillamide A can improve the inhibitory activity, as the inhibition effects of compounds 13c and 13o were 68.98% and 64.35%, respectively. Meanwhile, introduction of styryl, 2-methylpropenyl, and acetic acid methyl groups into the derivatives with m-phenylenediamine as the linker were beneficial to the inhibitory activity against the JAK/STAT signaling pathway, while the inhibition effects of compounds 14d, 14g and 14h were 62.98%, 89.70% and 71.30%, respectively.
These novel Neobacillamide A derivatives were also evaluated for their capability to inhibit the proliferation of A549 cell (Fig. 4). Intriguingly, the results showed that all these compounds displayed weak cytotoxicity (Table 1) at 25 μmol L−1 concentration. JAK/STAT signaling pathway is related to inflammation. Therefore, these Neobacillamide A derivatives with JAK/STAT inhibition can be used for further studies of anti-inflammatory activity.

|
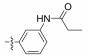
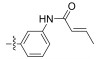
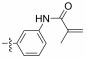
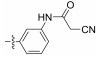
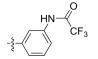
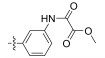
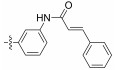
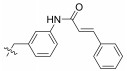
Fig. 4 Structure of the Neobacillamide A derivatives. |
|
|
Table 1 A549 cell inhibition and JAK/STAT inhibitory activities of Neobacillamide A derivatives 13b – 13p and 14a – 14i |
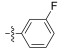
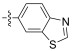
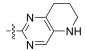
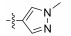
In conclusion, a series of Neobacillamide A derivatives were designed and synthesized. They were further evaluated for their effects on A549 cell, as well as their JAK/ STAT inhibitory activities. Five of these compounds displayed good JAK/STAT signal pathway inhibitory activity at a concentration of 25 μmol L−1 (all inhibitory potency were above 60%). The most promising compound, 14g, exhibited the highest inhibitory activity, with a 89.7% inhibition. Our further studies will evaluate the functions of these 5 marine alkaloid Neobacillamide A derivatives on JAK kinases.
AcknowledgementsThe authors are grateful for financial supports granted by the National Natural Science Foundation of China (Nos. 82073759 and 82003583), the Fund of Greater Bay Area Institute of Precision Medicine (Guangzhou) (No. IPM20 21C009), and the National Science and Technology Major Project for Significant New Drugs Development (No. 20 18ZX09735004).
Bahekar, R., Panchal, N., Soman, S., Desai, J., Patel, D., and Argade, A., et al., 2020. Discovery of diaminopyrimidine-carbo-xamide derivatives as JAK3 inhibitors. Bioorganic & Medicinal Chemistry Letters, 99: 103851. (  0) 0) |
Barua, S., Chung, J. I., Kim, A. Y., Lee, S. Y., Lee, S. H., and Baik, E. J., 2017. Jak kinase 3 signaling in microgliogenesis from the spinal nestin+ progenitors in both development and response to injury. Neuroreport, 28: 929-935. DOI:10.1097/WNR.0000000000000854 ( 0) |
Bhide, R. S., Keon, A., Weigelt, C., Sack, J. S., Schmidt, R. J., Lin, S., et al., 2017. Discovery and structure-based design of 4, 6-diaminonicotinamides as potent and selective IRAK4 inhibitors. Bioorganic & Medicinal Chemistry Letters, 27: 4908-4913. ( 0) |
Blunt, M. D., Koehrer, S., Dobson, R. C., Larrayoz, M., Wilmore, S., and Hayman, A., 2017. The dual Syk/JAK inhibitor cerdulatinib antagonizes B-cell receptor and microenvironmental signaling in chronic lymphocytic leukemia. Clinical Cancer Research, 23: 2313-2324. DOI:10.1158/1078-0432.CCR-16-1662 ( 0) |
Chen, M., Cheng, A., Chen, Y. Q., Hymel, A., Hanson, E. P., Kimmel, L., et al., 1997. The amino terminus of JAK3 is necessary and sufficient for binding to the common g chain and confers the ability to transmit interleukin 2-mediated signals. Proceedings of the National Academy of Sciences, 94: 6910-6915. DOI:10.1073/pnas.94.13.6910 ( 0) |
Clark, J. D., Flanagan, M. E., and Telliez, J. B., 2014. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. Journal of Medicinal Chemistry, 57: 5023-5038. DOI:10.1021/jm401490p ( 0) |
Clere-Jehl, R., Mariotte, A., Meziani, F., Bahram, S., Georgel, P., and Helms, J., 2020. JAK-STAT targeting offers novel therapeutic opportunities in sepsis. Trends in Molecular Medicine, 26: 987-1002. DOI:10.1016/j.molmed.2020.06.007 ( 0) |
Coffey, G. P., Feng, J., Betz, A., Pandey, A., Birrell, M., Leeds, J. M., et al., 2019. Cerdulatinib pharmacodynamics and relationships to tumor response following oral dosing in patients with relapsed/refractory B-cell malignancies. Clinical Cancer Research, 25: 1174-1184. DOI:10.1158/1078-0432.CCR-18-1047 ( 0) |
Elliott, N. E., Cleveland, S. M., Grann, V., Janik, J., Waldmann, T. A., and Dave, U. P., 2011. FERM domain mutations induce gain of function in JAK3 in adult T-cell leukemia/lymphoma. Blood, 118: 3911-3921. ( 0) |
Flanagan, M. E., Blumenkopf, T. A., Brissette, W. H., Brown, M. F., Casavant J. M., Shang-Poa, C., et al., 2010. Discovery of CP-690, 550: A potent and selective Janus kinase (JAK) inhibitor for the treatment of autoimmune diseases and organ transplant rejection. Journal of Medicinal Chemistry, 53: 8468-8484. DOI:10.1021/jm1004286 ( 0) |
Fleischmann, R., 2012. Novel small-molecular therapeutics for rheumatoid arthritis. Current Opinion in Rheumatology, 24: 335-341. DOI:10.1097/BOR.0b013e32835190ef ( 0) |
Harrison, C., Kiladjian, J. J., Al-Ali, H. K., Gisslinger, H., Waltzman, R., Stalbovskaya, V., et al., 2012. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. The New England Journal of Medicine, 366: 787-798. DOI:10.1056/NEJMoa1110556 ( 0) |
Hart, S., Goh, K. C., Novotny-Diermayr, V., Tan, Y. C., Madan, B., Amalini, C., et al., 2011. Pacritinib (SB1518), a JAK2/FLT3 inhibitor for the treatment of acute myeloid leukemia. Blood Cancer Journal, 1: e44. DOI:10.1038/bcj.2011.43 ( 0) |
Ito, M., Yamazaki, S., Yamagami, K., Kuno, M., Morita, Y., Okuma, K., et al., 2017. A novel JAK inhibitor, peficitinib, demonstrates potent efficacy in a rat adjuvant-induced arthritis model. Journal of Pharmacological Sciences, 133: 25-33. DOI:10.1016/j.jphs.2016.12.001 ( 0) |
Jensen, K. V., Cseh, O., Aman, A., Weiss, S., and Luchman, H. A., 2017. The JAK2/STAT3 inhibitor pacritinib effectively inhibits patient-derived GBM brain tumor initiating cells in vitro and when used in combination with temozolomide increases survival in an orthotopic xenograft model. PLoS One, 12: e0189670. DOI:10.1371/journal.pone.0189670 ( 0) |
Liu, Q., Sabnis, Y., Zhao, Z., Zhang, T., Buhrlage, S. J., Jones, L. H., et al., 2013. Developing irreversible inhibitors of the protein kinase cysteinome. Chemistry & Biology, 20: 146-159. ( 0) |
Lu, Z., Hong, C. C., Jark, P. C., Assumpção, A. L. F. V., Bollig, N., Kong, G., et al., 2017. JAK1/2 inhibitors AZD1480 and CYT387 inhibit canine B-cell lymphoma growth by increasing apoptosis and disrupting cell proliferation. Journal of Veterinary Medical Science, 31: 1804-1815. ( 0) |
Martínez, V., and Davyt, D., 2013. Total syntheses of bacillamide C and neobacillamide A; revision of their absolute configurations. Tetrahedron: Asymmetry, 24: 1572-1575. DOI:10.1016/j.tetasy.2013.11.001 ( 0) |
Monaghan, K. A., Khong, T., Burns, C. J., and Spencer, A., 2011. The novel JAK inhibitor CYT387 suppresses multiple signalling pathways, prevents proliferation and induces apoptosis in phenotypically diverse myeloma cells. Leukemia, 25: 1891-1899. DOI:10.1038/leu.2011.175 ( 0) |
O'Shea, J. J., Holland, S. M., and Staudt, L. M., 2013. JAKs and STATs in immunity, immunodeficiency, and cancer. The New England Journal of Medicine, 368: 161-170. DOI:10.1056/NEJMra1202117 ( 0) |
Changelian, P. S., Flanagan, M. E., Ball, D. J., Kent, C. R., Magnuson, K. S., and Martin, W. H., et al., 2003. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science, 302: 875-878. DOI:10.1126/science.1087061 ( 0) |
Schwartz, D. M., Bonelli, M., Gadina, M., and O'Shea, J. J., 2016. Type Ⅰ/Ⅱ cytokines, JAKs, and new strategies for treating auto-immune disease. Nature Reviews Rheumatology, 12: 25-36. DOI:10.1038/nrrheum.2015.167 ( 0) |
Shinde, P., Banerjee, P., and Mandhare, A., 2019. Marine natural products as source of new drugs: A patent review. Expert Opinion on Therapeutic Patents, 29: 283-309. DOI:10.1080/13543776.2019.1598972 ( 0) |
Thorarensen, A., Dowty, M. E., Banker, M. E., Juba, B., Jussif, J., Lin, T., et al., 2017. Design of a Janus kinase 3 (JAK3) specific inhibitor 1-((2S, 5R)-5-((7H-pyrrolo[2, 3-d]pyrimidin-4-yl) amino)-2-methylpiperidin-1-yl) prop-2-en-1-one (PF-06651600) allowing for the interrogation of JAK3 signaling in humans. Journal of Medicinal Chemistry, 60: 1971-1993. DOI:10.1021/acs.jmedchem.6b01694 ( 0) |
Verbsky, J. W., Bach, E. A., Fang, Y. F., Yang, L. P., Randolph, D. A., and Fields, L. E., 1996. Expression of Janus knase 3 in human endothelial and other non-lymphoid and non-myeloid cells. Journal of Biological Chemistry, 271: 13976-13980. DOI:10.1074/jbc.271.24.13976 ( 0) |
West, K., 2009. CP-690550, a JAK3 inhibitor as an immunosup-pressant for the treatment of rheumatoid arthritis, transplant rejection, psoriasis and other immune-mediated disorders. Current Opinion in Investigational Drugs, 10: 491-504. ( 0) |
Yin, Y., Chen, C. J., Yu, R. N., Shu, L., Wang, Z. J., Zhang, T. T., et al., 2020. Novel 1H-pyrazolo[3, 4-d] pyrimidin-6-amino deri-vatives as potent selective Janus kinase 3 (JAK3) inhibitors. Evaluation of their improved effect for the treatment of rheu-matoid arthritis. Bioorganic Chemistry, 98: 103720. DOI:10.1016/j.bioorg.2020.103720 ( 0) |
Yu, L. L., Li, Z. Y., Peng, C. S., Li, Z. Y., and Guo, Y. W., 2009. Erratum: Neobacillamide A, a novel thiazolecontaining alka-loid from the marine bacterium Bacillus vallismortis C89, as-sociated with South China Sea sponge Dysidea avara. Helvetica Chimica Acta, 92(3): 607-612. DOI:10.1002/hlca.200800349 ( 0) |