 2009, Vol. 25
2009, Vol. 25


2. 北京协和医学院中国医学科学院比较医学中心;
3. 北京协和医学院中国医学科学院中法合作艾滋病研究中心;
4. 法国发展研究院蒙特利尔大学UMR145研究所;
5. 巴黎第五大学肿瘤疫苗免疫治疗中心病毒分子研究所
海南省自1991年发现首例人类免疫缺陷病毒(HIV)感染者以来,已蔓延至全省13个市县,为了解感染者之间的传播模式,于2007年5月采集全省13个市县的88例HIV感染者的血液标本进行HIV基因系列分析,以了解HIV毒株亚型分布及基因变异情况,为艾滋病(AIDS)预防和控制提供科学依据。现将结果报告如下。
1 材料与方法 1.1 材料标本来源于2007年海南省有艾滋病疫情的市县采集艾滋病病毒感染者和病人(HIV/AIDS)96份血液样品,其中88份为HIV阳性者标本,8份为阳性者配偶的阴性标本。其中男性72例,女性16例,平均年龄31岁,静脉吸毒56例,性传播19例,本省户籍84例,外省户籍4例。
1.2 方法从血浆中提取病毒RNA;逆转录病毒的RNA的聚合酶(pol)和膜糖蛋白基因(env)片断;巢式PCR扩增env V3~V5片断,目的片段大小为660 bp;巢式PCR扩增pol片断,覆盖HIV-1蛋白酶和逆转录酶的编码区,目的片段大小为1860 bp。
1.3 序列测定 1.3.1 pol片段的测序引物pol片段大小约为1860 bp,采用分6个引物分别测序,再使用DNAStar Lasergene 7.1的Se-qMan软件拼接得到目的片段。引物如下: AV150: 5′-GTGGAA AGG AAG GAC ACC AAA TGA AAG-3′;POLM1bis: 5′-CAG GAA TGG ATG GCC CAA-3′;POLM2bis: 5′-CAC AGG GAT GGA AAG GAT CACC-3′;POLM8: 5′-CTG TAT ATC ATT GAC AGT CCA G-3′;POLM9: 5′-ATT GAA CTT CCC AGA AGT CTT GAG TT-3′;POLM4: 5′-CTA TTA GCT GCC CCA TCT ACATA-3′。
1.3.2 env片段的测序引物env片段大小约为660 bp,采用正反引物双向测序,再拼接的方法得到目的片段。引物如下: ES7 (正向) : 5′-CTC AAC TGC TGT TAA ATG GCA GTC TAGC-3′;ES8 (反向) : 5′-TAT AAT TCA CTT CTC CAA TTG TCCCTC A-3′。
1.4 序列分析使用Blast程序将测序所得结果与GenBank中的序列进行比较,检查在实验过程中是否有交叉污染;在Genebank (NCBI)或HIV Database (Los Alamos)中检索所有独立的HIV-1亚型的基因型(A1,A2,B,B',C,D,F1,F2,G,H,J,K,L),以及相关的重组基因型(CRF01-AE, CRF02 _ AG, CRF07_BC, CRF08 _BC, CRF13 _cpx, CRF15_01B, CRF33_ 01B, CRF34_01B)的核酸序列;使用ClustalX 1.83对所有正确的序列以及在基因库中检索出来的序列共同进行比对。根据最大似然法(ML)的计算结果,判断分析在所有HIV-1阳性感染者之间有无传播链的存在。判断标准为pol基因序列的计算结果支持率 > 98%,分支长度 < 0.015。
2 结果 2.1 HIV分型及地区分布经pol基因及env基因序列比对分析,88份样品中有83份样品可确定分为6种HIV-1亚型或重组亚型,另外5份样品未能扩增出全部pol基因和env基因片段,无法明确分型。除五指山市外,全部有艾滋病疫情的采样点市县均有CRF01-AE亚型存在,3个市县有2种亚型,2个市县有3种亚型。
2.2 感染途径与亚型的关系(表 1)| 表 1 83份样品中HIV-1亚型在不同感染途径的人群中分布 |
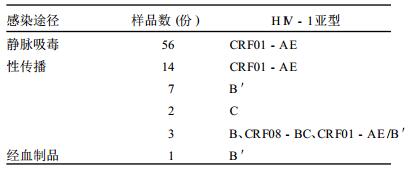
83份样品的感染途径分为静脉吸毒、性传播和经血液制品3类。静脉吸毒人群感染的HIV-1亚型全部为CRF01-AE亚型,未发现其他亚型。经性传播感染的人群亚型复杂多样,包括检出的全部亚型。
2.3 系统进化树及传播群 2.3.1 pol基因将所获得的83份样品基因序列与各亚型地区或国际参考株pol基因序列构建系统进化树。在83个样品中,有66个样品(79.5%)分属于4个大小不同的传播群(transmission clusters),它们都是根据最大似然法的计算结果(bootstrap值是在重复1 000次基础上得到),以计算结果支持率(bootstrap values) > 98%,平均分支长度(average branch lengths)即遗传距离(mean genetic distance) < 0.015为判断标准。传播群1最大,有59例,形成了一个庞大的传播群;传播群2有3人,传播群3和4各有2人。非传播群有17个样品(20.5%)。传播群1有53人为静脉吸毒人群,有6人为经异性性传播,其中有4人为吸毒者的配偶。传播群2均为吸毒人员。传播群3,其性传播感染可能性较大。传播群4为异性性传播感染。非传播群的样品除1例为经血制品感染HIV外,其他均为性传播途径感染。
2.3.2 env基因将所获得的83份样品基因序列与各亚型地区或国际参考株env基因序列构建系统进化树。pol序列的所有传播群在env序列都有体现,但其分支长度(遗传距离)均较pol序列的传播群长,均值都 > 0.015。在分析pol序列得到的4个传播群当中,传播群2,3,4的个体没有变化,且传播群2,4的bootstrap值仍然 > 98%,而传播群3的bootstrap值较低 < 90%;传播群1由原来的59人增加到61人,bootstrap值仍然 > 98%。
3 讨论本次研究结果表明,海南省居首位的HIV亚型为CRF-AE亚型(84.33%,70/83),同时,新发现1例CRF01-AE/B′重组亚型。从HIV-1亚型的地域分布来看,不同亚型在呈不平衡分布,CRF01-AE亚型HIV-1流行区域最广,除五指山市外见于所有调查市县,其次为B′亚型。在海南省西南地区(三亚市21例、乐东县13例、东方市12例和儋州市11例) CRF01-AE亚型占绝对优势,占全部被调查市县的68.6%。吸毒者的CRF01-AE亚型分布在西南地区和沿海的万宁市与临高县。经性传播的亚型散在分布在北部各市县,呈现出从北部向中部山区和全省范围蔓延扩散的趋势。
本次研究结果表明,海南省吸毒人群均为CRF01-AE重组亚型(100%),未发现其他亚型。其不同于国内其他省份的CRF-BC亚型的原因,可能CRF-BC亚型在海南省的流行还处于较低水平(1例),其向吸毒人群扩散慢于其他亚型;也可能与亚型的先期基础效应有关〔1〕,CRF01-AE进入吸毒人群后就快速传播开来。经性传播人群的亚型呈多样性,包括检出的全部亚型,与内陆经性传播亚型呈多样性的特点相似。对于病毒
基因序列系统发育分析的方法最近已被用来分析各地急、慢性HIV-1感染人群内直接或间接的传播链〔2-6〕。发现这一方法能够在分子水平追踪HIV-1的传播链。本研究ML进化树显示,近80%(66/83)感染者可确定其流行病学关系,对于传播群1,其感染HIV的来源很可能是1~2人,然后通过共用针头静脉吸毒的方式逐渐扩散开,形成了一个奠基者效应(Founder Effect)。在进化树上他们关系很近共同形成了一个很密集的分支群。同样,也表明传播群1、传播群2和非传播群的CRF01-AE亚型的进化差异,非传播群的CRF01-AE亚型有8人,均为异性性传播感染HIV,与传播群2以及福建省和广西壮族自治区已知的毒株的基因距离更为接近,而与传播群1距离较远。表明海南省的CRF01-AE毒株大体上来源于2个途径,最主要的是静脉吸毒以及吸毒者相关的性传播,其次还有少量的CRF01-AE来源于性传播,这个性传播很可能是CRF01-AE从邻近的省份扩散而来。
(致谢:北京协和医学院中国医学科学院比较医学中心各位老师的支持与配合和海南省各市县疾病预防控制中心的协助)| [1] | Thomson MM, Pérez-A lvarez L, Nàjera R. Molecular epidemiology of HIV-1 genetic forms and its significance for vaccine deve-lopment and therapy[J]. Lancet Infect Dis, 2002, 2(8) : 461–471. DOI:10.1016/S1473-3099(02)00343-2 |
| [2] | Bao L, Vidal N, Fang H, et al. Molecular tracing of sexual HIV type 1 transmission in the southwest border of China[J]. AIDS Research and Human Retroviruses, 2008, 24(5) : 733–742. DOI:10.1089/aid.2007.0269 |
| [3] | Hue S, Clewley JP, Cane PA, et al. HIV-1 pol gene variation is suffi-cient for reconstruction of transmissions in the era of antiretroviral therapy[J]. AIDS, 2004, 18 : 719–728. DOI:10.1097/00002030-200403260-00002 |
| [4] | Pao D, Fisher M, Hue S, et al. Transmission of HIV-1 during primary infection:relationship to sexual risk and sexually transmitted infec-tions[J]. AIDS, 2005, 19 : 85–90. DOI:10.1097/00002030-200501030-00010 |
| [5] | Hue S, Pillay D, Clewley JP, et al. Genetic analysis reveals the com-p lex structure of HIV-1 transmission within defined risk groups[J]. Proc Natl Acad Sci, 2005, 102 : 4425–4429. DOI:10.1073/pnas.0407534102 |
| [6] | 谭晓华, 狄春红, 杨磊. 病毒微生物对HIV-1感染抑制作用研究进展[J]. 中国公共卫生, 2008, 24(12) : 1423. |