


甲壳素, 又名几丁质、壳多糖, 其化学本质为聚-N-乙酰-D-氨基葡萄糖, 是甲壳类动物外壳、节肢动物表皮、低等动物细胞膜、高等植物细胞壁等生物组织中广泛存在的一种天然动物纤维素。甲壳质脱乙酰化, 得到能溶于稀有机酸的物质, 即壳聚糖(Chitosan)。甲壳质脱乙酰化后, 溶解性能提高, 能被人体吸收。脱乙酰甲壳质经辐射降解生成低聚壳聚糖, 能溶于水, 更易于吸收。有实验证明, 低聚壳聚糖对辐射有明显的抑制能力[1, 2]。本课题通过整体动物的脾脏对低聚壳聚糖抗辐射的作用进行了实验观察。
1 材料与方法 1.1 低聚壳聚糖本实验研究的低聚壳聚糖由本院制备, 以食品级大分子壳聚糖为原料, 敏化剂协同作用, 经辐照降解氧化后制备而成。分子量<2 kD, 分散系数1.21。灌胃前用蒸馏水配制成溶液。
1.2 实验动物选用6周龄, 雄性, 清洁级SX1近交系小鼠, 由山西省肿瘤研究所动物实验室提供, 许可证号: SCXK(晋) 2007-001。中国辐射防护研究院GLP中心动物实验室饲养管理。
1.3 辐射源60Coγ源, 中国辐射防护研究院附属医院放疗科远距离治疗机, 活度4.07×1013Bq(1 100Ci)。
1.4 主要试剂beta-actin抗: sc-47778, Santa Cruz Biotechnology, 美国; 小鼠来源的P53抗体: sc-73566, Santa Cruz Biotechnology, 美国; 兔来源的Anti-Gadd45抗体: bs-1360R, 北京博奥森生物技术有限公司。
1.5 试验方法 1.5.1 脾脏器系数实验实验小鼠随机分为正常对照组、单纯照射组、低聚壳聚糖组(300mg/kg) 3个实验组, 每组40只。连续灌服低聚壳聚糖14d, 1次/d。正常对照组和单纯照射组灌服蒸馏水。给药第14天除正常对照组外其余组小鼠均接受7 Gy 60Coγ射线全身照射, 吸收剂量率为0.33 Gy/min, 照射误差1%。在第5、10、15、20、30天取5只活存动物脱颈处死, 取脾脏, 称取脾脏重量。
1.5.2 脾脏凋亡基因检测实验小鼠随机分为正常对照组、单纯照射组、低聚壳聚糖组(300mg/kg) 3个实验组, 每组10只。连续灌服低聚壳聚糖14d, 1次/d。正常对照组和单纯照射组灌服蒸馏水。给药第14天除正常对照组外其余组小鼠均接受5 Gy 60Coγ射线全身照射, 吸收剂量率为0.33 Gy/min, 照射误差1%。照射后6h, 摘取动物脾脏, 采用RT-PCR方法检测bax、Bcl-2、caspase-3, 步骤如下:总RNA提取-RT(逆转录)-PCR(聚合酶链反应)-琼脂糖凝胶电泳。elDoc2000型凝胶成像系统观察结果, 产物带的密度用凝胶扫描系统分析记录结果, 以GAPDH为参照, Photoshop7.0分析条带。各基因RT-PCR试验条件见表 1。
|
|
表 1 各基因RT-PCR试验条件一览表 |
上述方法摘取的脾脏采用western手段检测P53、GADD45蛋白。大致步骤如下:蛋白样品的制备-SDS-PAGE-转膜-封闭-一抗孵育-二抗孵育-ECL底物化学发光显色, 扫描。
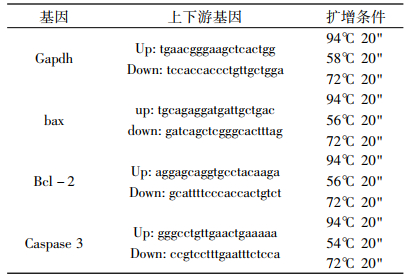
2 结果 2.1 脾脏器系数结果小鼠在照射后5、10、15、20、30d的脾脏器系数结果见表 2。由表 2可见, 单纯照射组在照射后第5、10d脾脏器系数最小, 与正常对照组相比降低最多, 仅为正常对照组的16%, 在第15、20天脾脏器系数有所恢复, 在第30天脾脏器系数与正常对照组相差最小, 为正常对照组的55%。正常对照组与单纯照射组相应时间的脾脏器系数有统计学差异。低聚壳聚糖组照射后第5 ~ 20天, 各时间的脾脏器系数值有缓慢上升趋势, 为正常对照组的47% ~ 61%, 第30天的脾脏器系数为正常对照组的98.7%, 各时间点脾脏器系数与单纯照射组相应时间的脾脏器系数有统计学差异。
|
|
表 2 7Gy照射后不同时间小鼠脾脏器系数比较 |
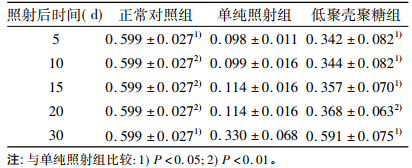
表 3结果显示, 动物受到照射后, 凋亡基因Bax、Bcl-2、Caspas-3表达变化不同。单纯照射组相关凋亡基因与正常对照组相比bax上调, bc1-2上调, caspas-3上调, bcl-2/bax比值减少, 低于1;低聚壳聚糖组凋亡相关基因与正常对照组相比caspas-3下调, bax上调, bc1-2上调, bcl-2/bax比值提高, 比值高于1, 且与单纯照射组有统计学差异。
|
|
表 3 5Gy照射动物后6h脾脏凋亡基因变化情况 |
图 2、3显示, 动物受到照射后, 单纯照射组脾脏组织P53蛋白、Gadd45蛋白较正常对照组表达提高, 低聚壳聚糖组的蛋白表达较单纯照射组降低。

|
图 1 WESTERN方法检测P53、Gadd45蛋白结果 |

|
图 2 低聚壳聚糖对受照动物脾脏P53蛋白表达的影响 |

|
图 3 低聚壳聚糖对受照动物脾脏Gadd45蛋白表达的影响 |
本实验研究整体动物受照后脾脏的脏器系数、脾脏凋亡基因和辐射敏感蛋白的变化, 探讨受照动物脾脏变化规律及研究其分子原因, 同时研究低聚壳聚糖对辐射损伤保护的作用机理。动物受到7Gy照射后, 在照后第3天, 白细胞下降至最低点, 到第7 ~ 10天开始恢复, 这个时段动物代偿, 辐射损伤最大, 死亡数量较多, 脾脏器系数下降最明显, 而在第15 ~ 20d, 由于脾脏的再生作用, 脏器系数有所提高。而低聚壳聚糖组动物脾脏器系数在照后20d内变化不大, 而且到30d时, 脾脏系数接近正常对照组。照射早期, 大量细胞凋亡可能是导致脾脏细胞数减少的重要原因, 继而引起后期免疫功能的受损。细胞凋亡是影响细胞辐射敏感性的重要事件。Bcl-2和Bax是近年来发现的一对关系密切的凋亡基因, 在细胞凋亡调控中起重要作用。Bcl-2抑制细胞的凋亡, Bax则促进细胞的凋亡。Bcl-2基因是较强的抗凋亡作用基因, 能够抑制或阻断多种因素引起的细胞凋亡。大剂量照射后, 它的抗凋亡作用受到抑制甚至失去了抑制细胞凋亡的作用, 结果使大量细胞出现凋亡, 导致单纯照射组Bcl-2表达升高。Bcl-2/bax比例对决定细胞命运起着关键的作用。当bax二聚体形成时, 便诱导细胞凋亡。而bcl-2表达量上升, Bax二聚体分开, 与Bcl-2形成稳定的Bcl-2/bax异源二聚体以及Bcl-2-Bcl-2同源二聚体时, 则中和了Bax二聚体诱导细胞凋亡的作用。当Bcl-2/Bax比值大于1时, 细胞凋亡无明显增加; 而Bcl-2/Bax比值小于1时, 细胞凋亡多明显增加。本实验低聚壳聚糖组抑制促凋亡基因Bax、caspas-3表达, 促进抑制凋亡基因Bcl-2表达, Bcl-2/ Bax比值大于1, 而且与单纯照射组有统计学差异。减少细胞的凋亡, 保持细胞存活是辐射损伤防治措施的根本所在。结果表明低聚壳聚糖有抑制辐射损伤动物的凋亡作用。
P53在损伤存活中起重要作用并因此被誉为"基因警察"。P53不仅可以通过活化P21和Gadd45引起细胞G1期阻滞和DNA修复, 还可调节细胞的增殖与凋亡。研究发现:大剂量电离辐射诱导细胞凋亡的过程中P53基因蛋白表达显著增加, 说明P53参与辐射诱导的细胞凋亡。本实验单纯照射组P53蛋白增加, 与上述研究一致, 而低聚壳聚糖组P53蛋白表达降低, 说明低聚壳聚糖组对P53蛋白表达有抑制作用。
Gadd45是一个生长阻滞和DNA损伤基因, 也是第一个被检出的P53下游靶基因。Gadd45可以通过P53依赖及非依赖两条途径被诱导表达增高, 参与细胞周期检查点、细胞凋亡、DNA损伤修复以及信号传导等重要细胞生命活动的调节[3, 4]。在电离辐射的作用下, Gadd45可以通过P53依赖的方式表达上调。研究表明, 高剂量X射线引起P53蛋白增多可诱导Gadd45增加[5], 本实验单纯照射组Gadd45、P53蛋白表达升高, 与研究结果一致。但低聚壳聚糖组Gadd45、P53蛋白表达有降低趋势。结果说明, 低聚壳聚糖对动物的辐射敏感蛋白的保护作用。
| [1] |
Nishimura Y, Kim HS, Ikota N, et al. Radioprotective effect of chitosan in sub-lethally X-ray irradiated mice[J]. J Radiat Res (Tokyo), 2003, 44(1): 53-8. DOI:10.1269/jrr.44.53 |
| [2] |
Ilín LA, Andrianova IE, Glushkov VA. Dependence of radioprotective activity of chitosan on its molecular mass[J]. Radiats Biol Radioecol, 2004, 44(2): 176-8. |
| [3] |
Kastan MB, Zhan Q, El-Deity WS, et al. A mammalian cell cycle checkpoint pathmay utilizing p53 and defective in ataxia-telangiectasia[J]. Cell, 1992, 71(4): 587-597. DOI:10.1016/0092-8674(92)90593-2 |
| [4] |
Gujuluva CN, Back JH, Shin KH, et al. Effect of UV-irradiation on cell cycle, viability and the expression of p53 and gadd 45 genes in normal and HPV-immortlized human oral keratinocytes[J]. Oncogene, 1994, 9(7): 1 819-1 827. |
| [5] |
牟颖, 刘树铮, 刘建香. X射线全身照射对小鼠胸腺细胞GADD45及Rb蛋白表达的影响[J]. 中华放射医学与防护杂志, 1998, 18(6): 408-409. DOI:10.3760/cma.j.issn.0254-5098.1998.06.013 |