
Clerodane diterpenoid glycosides from the tuberous roots of Paratinospora sagittata: targeted isolation, structure characterization and immunomodulatory properties

Abstract
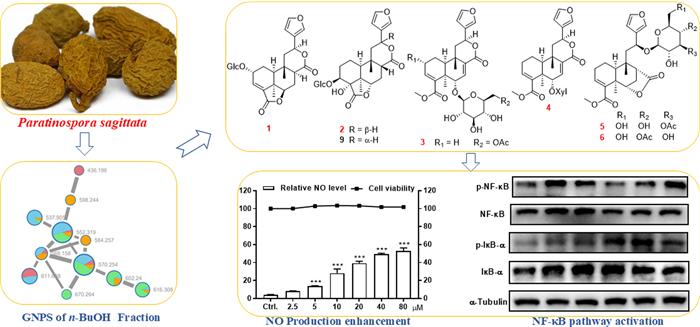
Guided by the MS/MS-based molecular networking, eight previously undescribed clerodane diterpenoid glycosides, designated tinospinosides F–M (1−8), along with 12 known analogues (9−20), were isolated from the tuberous roots of Paratinospora sagittata. Structural elucidation of the undescribed compounds was achieved through comprehensive spectroscopic analyses (NMR, HRESIMS), with their absolute configurations confirmed via single-crystal X-ray diffraction, TD-DFT/ECD computational analyses, and chemical degradation. Immunomodulation evaluation on all the isolates revealed that compounds 6 and 7 exerted significant promoting effect toward NO production in RAW264.7 macrophages. Further study demonstrated that 6 could enhance the release of immune cytokines (e.g., TNF-α) and upregulate the protein expression of iNOS and COX-2, which was potentially mediated through the activation of NF-κB signaling pathway.Graphical Abstract
Keywords
Paratinospora sagittata Clerodane Diterpenoid glycosides Absolute configuration Immunomodulatory1 Introduction
The genera Paratinospora and Tinospora, two closely related genera in the family Menispermaceae, are predominantly found in the tropical and subtropical regions of Asia, Australia, and Africa [1–3]. Before 2025, plants of genus Paratinospora (P. sagittata and P. dentata) belong to the genus Tinospora, while Wei Wang et al. formally established the new genus Paratinospora based on a combined analysis of plastid and nuclear DNA sequence data [4, 5]. Medicinal plants in the Paratinospora/Tinospora genera are extensively employed as folk medicine in countries of Southeast Asia as immunomodulatory agents [6–8]. Bioactive constituents in genera Paratinospora/Tinospora such as alkaloids and diterpenoids also demonstrate significant activities including anti-inflammatory, antimicrobial, antiviral and antioxidant effects [1–3]. Paratinospora sagittata (Oliv.) Wei Wang, commonly known as "Jin Guo Lan" in traditional Chinese medicine, holds a prominent position in the Chinese Pharmacopeia [9]. Its rhizomes are extensively utilized for their therapeutic potential in fever reduction, detoxification, alleviation of swelling and pain management [10]. Previous studies on the chemical constituents of this species have primarily focused on the EtOAc soluble part of its EtOH extract, with the main components identified as diterpenoids [11–15]. To the best of our knowledge, the n-BuOH soluble part has not been investigated before.
As a continuation of our research on diterpenoids from family Menispermaceae [16–18], the current study employed a targeted isolation using MS/MS-based molecular networking analysis, which revealed characteristic signatures of clerodane diterpenoid glycosides within the n-BuOH soluble part of P. sagittata. This approach led to the isolation and identification of 20 clerodane diterpenoid glycosides, including eight previously undescribed compounds. The structures of the undescribed compounds were determined through extensive spectroscopic analyses, with their absolute configurations further confirmed by single-crystal X-ray diffraction, TD-DFT/ECD computational method, and chemical methods. The immunomodulatory effects of all isolated compounds were assessed in RAW264.7 cells. This study details the isolation, structure elucidation, and the observed immunomodulatory activities of these compounds.
2 Results and discussion
2.1 Molecular networking analysis
Tuberous roots of P. sagittata (30 kg) were powdered and then extracted with 95% EtOH at room temperature to give a residue, which was suspended in H2O and then partitioned successively with EtOAc and n-BuOH. The n-BuOH partition was further separated into four fractions (A−D) by D101 macroporous resin and silica gel column chromatographies. Subsequently, HPLC-HRESIMS/MS data of the four fractions were uploaded to Global Natural Products Social Molecular Networking database to construct a molecular network, which revealed three major clusters and several minor clusters. According to the previous recognition of the MS/MS data of clerodane diterpenoids, the diagnostic ions are the fragment ions at m/z 377 → m/z 317 → m/z 299 [18]. Then, the fragment ions at m/z 317 → 299 in the MS–MS data of one cluster in the n-BuOH fraction suggested the presence of clerodane diterpenoids, which primarily presented in fractions B and C (see Fig. 1). Subsequently, further in-depth investigation led to the isolation of eight undescribed clerodane diterpenoids and 12 known analogues (1−20, Fig. 2).
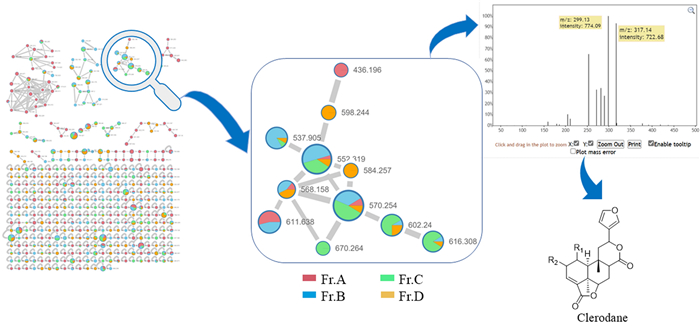
MS/MS-based molecular networking of the n-BuOH fraction of P. sagittate
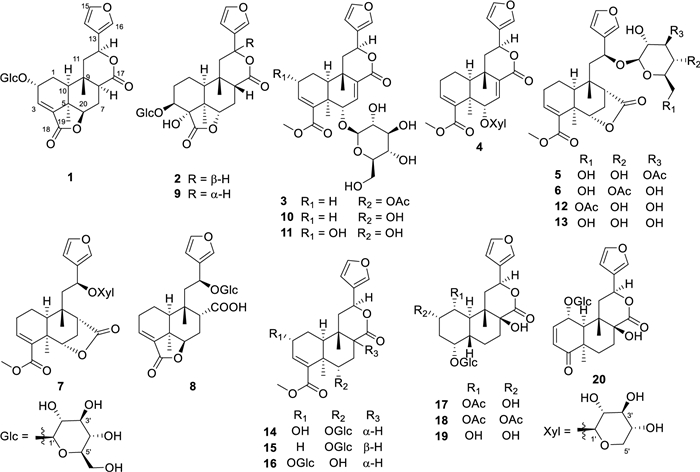
Chemical structures of compounds 1–20
2.2 Structure elucidation
Compound 1, a colorless gum, had the molecular formula of C26H32O11 established by HRESIMS (11 degrees of unsaturation). 1D NMR and HSQC data revealed signals for two ester carbonyl groups (δC 175.8 and 171.0), a β-substituted furan unit [δH 6.53 (1H, dd, J = 1.8, 0.7 Hz), 7.52 (1H, t, J = 1.7 Hz) and 7.61 (1H, m); δC 109.7, 125.4, 141.5 and 145.2], a trisubstituted double bond [δH 6.84 (1H, d, J = 3.6 Hz); δC 136.7 and 137.5], a β-glucose moiety [δH 3.18, 3.28, 3.33, 3.35, 3.67, 3.90 and 4.50 (1H, d, J = 7.8 Hz); δC 62.8, 71.8, 75.1, 78.0, 78.1 and 104.1] and two methyl groups [δH 0.94 and 1.39 (each 3H, s); δC 22.7 and 30.2]. The gross structure for 1 was accomplished by interpretation of 2D NMR data, especially HMBC and 1H−1H COSY data (Fig. 3). In the 1H−1H COSY spectrum, the cross peaks of H-10/H2-1/H-2/H-3 suggested the presence of the key fragment (C-10/C-1/C-2/C-3). Then, the HMBC correlations from protons of a tertiary methyl (H3-19) to C-4, C-5 and C-10 established the formation of ring A. Moreover, the 1H−1H COSY correlations of H-6/H2-7/H-8 and the HMBC correlations from H2-7 and H-10 to C-8 and C-9, together with the HMBC correlations from H3-20 to C-8, C-9 and C-10 constructed the six-membered ring B. The 1H − 1H COSY correlations from H2-11 to H-12 together with the HMBC correlations from H-14 and H-16 to C-12 attached the furan ring to C-12. Key HMBC correlation from H-1′ (δH 4.50) to C-2 suggested that the sugar moiety was attached to C-2. As nine of 11 degrees of unsaturation were accounted by the aforementioned furan ring, double bond, carbonyl groups, sugar moiety and the basic 6/6 bicyclic framework, the remaining two degrees of unsaturation required two additional rings in the structure of 1. Thus, the key HMBC correlation from H-12 to the ester carbonyl carbon at δC 174.6 (C-17) and H-6 to another ester carbonyl carbon at δC 171.0 (C-18) indicated the formation of 12,17-lactone bridge and 6,18-lactone bridge, respectively.
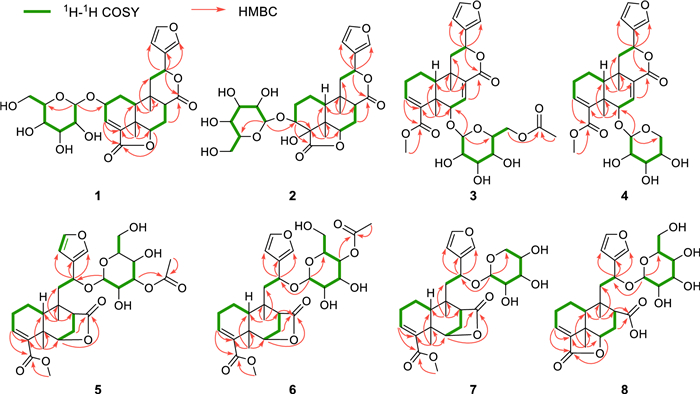
Selected 1H−1H COSY and HMBC cross-peaks for compounds 1−8
The relative configuration of 1 was mainly based on NOESY spectrum (Fig. 4). NOESY correlations of H3-19/H-6 and H-10, H-6/H-8 and H-7 at δH 2.47, H-8/H-10 and H-12, indicated their spatial proximity and a random determination of the α-orientation of these protons. Conversely, NOESY cross peaks between the H3-20 with H-2 and H-7 at δH 1.43, revealed the β-orientation for CH3-20 and H-2. The β-configuration of the anomeric proton was established by analyzing the coupling constant of the anomeric proton (J = 7.8 Hz). Subsequent acid hydrolysis confirmed that the retention time of the obtained product matched exactly with that of the standard D-glucose. Finally, the experimental ECD spectrum of 1 matched the calculated ECD spectrum of the (2R,5R,6R,8S,9S,10S,12S)-isomer (Fig. 5). Thus, the structure of 1 was fully characterized and was given the trivial name tinospinoside F, following tinospinosides A−E reported from the same species [19, 20].
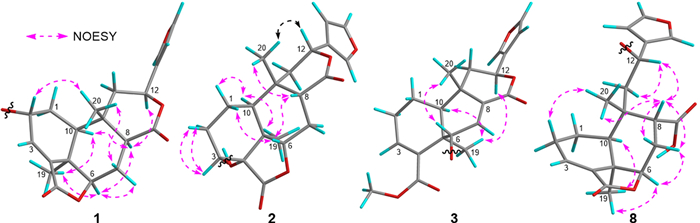
Key NOESY correlations for compounds 1−3 and 8
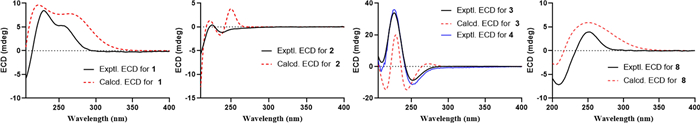
Experimental ECD spectra of 1−4 and 8 and calculated ECD spectra of 1−3 and 8
HRESIMS analysis of compound 2 revealed a quasi-molecular ion at m/z 556.2393 [M + NH4]+ (calcd for C26H38O12N+, 556.2389), indicative of a molecular formula of C26H34O12 and isomeric with borapetoside A (9) [21]. The 1D NMR data of 2 showed high similarity to those of 9 with the major differences being attributed to the chemical shift of H-12 (δH 5.63 in 2; δH 5.90 in 9), indicating that 2 was the 12-epimer of 9. Detailed 2D NMR data analyses further secured the planar structure of 2, which was the same as that of 9. The relative configuration of 2 was established mainly by NOESY experiments (Fig. 4) and 1D NMR data. The chiral centers in rings A and B were assigned to be the same as those in 9 based on the highly matched 1D NMR data. Key NOESY correlations of H3-20/H-12 and H-8 supported the orientation of H-12 as β. The absolute configuration of 2 was determined by comparison its experimental ECD curve with its theoretical one. Thus, the structure of compound 2 (tinospinoside G) was assigned.
Compound 3, a colorless gum, possessed a molecular formula of C29H32O12 by analysis of its HRESIMS data (m/z 577.2279 [M + H]+, calcd for C29H33O12+, 577.2280). The 1D NMR data and HSQC spectrum exhibited signals for an acetyl group [δH 2.17 (3H, s); δC 172.0 and 20.8], a β-substituted furan unit [δH 7.46 (1H, brs), 7.42 (1H, brs) and 6.42 (1H, brs); δC 143.7, 139.8, 123.7 and 108.6], two ester carbonyl groups (δC 171.6 and 168.7), two trisubstituted double bonds [δH 6.99 (1H, s), 6.49 (1H, t, J = 3.4 Hz); δC 138.4, 136.7, 136.7 and 136.2] and two methyl groups [δH 1.37 (3H, s) and 1.17 (3H, s); δC 27.9 and 22.3]. The aforementioned NMR data of 3 exhibited high resemblance with those of 10 (borapetoside F [22]), except for the additional signals of an acetyl group. In the HMBC spectrum, the cross-peak from H2-6′ (δH 4.39 and 4.31) to the carbonyl of acetyl group (δC 172.0) suggested that the 6′-OH in 10 was acetylated in 3. Detailed 2D NMR correlations including 1H-1H COSY and HMBC analysis supported that planar structure of 3 as depicted. The relative configuration of 3 was assigned the same as that of 10 by their highly-matched 1D NMR data. As shown in Fig. 5, the experimental ECD curve of 3 matched the calculated ECD spectrum of the (5R,6S,9S,10S,12S)-isomer. Thus, the structure of 3 was fully characterized and given the trivial name tinospinoside H.
Similar with compound 3, the NMR data of compound 4 (tinospinoside Ⅰ) also showed high resemblance with those of 10. Extensive comparison of the 1D NMR data of 4 with those of 10 revealed that the glucopyranose unit in 10 was replaced by a xylopyranose unit in 4. Further 2D NMR analysis supported the planar structure of 4 as shown in Fig. 3. The xylopyranose unit was determined as D-configuration by HPLC comparison with those of the standard ones. While the absolute configuration of the aglycone of 4 was the same as that of 3 by their highly-matched ECD curves.
Compounds 5 and 6 were assigned the same molecular formula C29H38O12 by HRESIMS analyses and their 1D NMR data showed high resemblance. With the aid of full 2D NMR experiments, the 1D NMR data of 5 and 6 were completely assigned and exhibited high similarity to those of tinopanoid T (12) [23], the co-isolated analogue in current research. A detailed comparison revealed that the key structural difference among these analogues was the site of acetylation. Whereas compound 12 is acetylated at the 6′-O position, compounds 5 and 6 are acetylated at the3′-O and 4′-O positions, respectively. These structural assignments were unambiguously supported by the chemical shift comparisons of H-3′, H-4′ and H-6′ in the three structures, as well as their HMBC correlations to the corresponding acetyl carbonyl carbons. The configurations of 5 and 6 were determined to be identical with those of 12 based on their consistent NMR coupling patterns, NOESY data and ECD curves. Their structures were further secured by chemical correlation with 12. The alkaline hydrolysis of 5, 6 and 12 generated the same product 13 (Fig. 6), which was verified by comparison of their NMR data. The absolute configuration of 12 was determined by single crystal X-ray diffraction [CCDC number: 2376200; Frack parameter = − 0.01 (10)]. Hence, the structures of 5 and 6 were established as depicted and given the trivial names tinospinosides J and K, respectively.
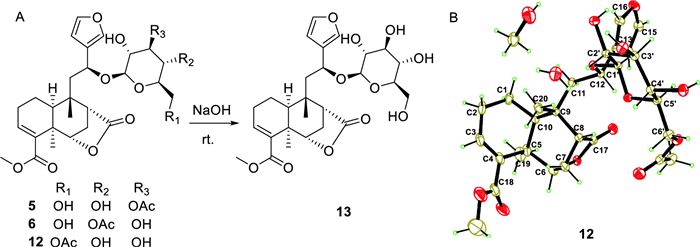
A Chemical transformation from 5, 6 and 12 to 13; B Ortep drawing of 12
Compound 7 was isolated as a colorless gum and had the molecular formula of C26H34O10 as determined by HRESIMS. The 1D NMR data of 7 bore a resemblance to those of 13 [24], except for the replacement of the glucopyranose unit in 13 by a xylopyranose unit in 7. This assignment was further confirmed by the 1H − 1H COSY correlations of H-1′/H-2′/H-3′/H-4′/H2-5′, together with the HMBC correlations of H-1′/C-12. The relative configuration of the aglycone of 7 was assigned to be the same as that of 13 by comparing their highly matched 1D NMR data. Especially, the configuration of H-12 was determined by the empirical rule established by Gao et al. [25], the coupling constants between H-12 and H-11 (J = 9.9, 3.3 Hz) suggested the relative configuration of H-12 was S*. The absolute configuration of 7 was determined by ECD comparison with those of 5 and 6, nearly identical cotton effects revealed that they have the same absolute configuration in the aglycone part. Thus, compound 7 was assigned as depicted and named tinospinoside L.
Compound 8, a colorless gum, had the molecular formula C26H34O11 as determined by the HRESIMS ion at m/z 545.1993 [M + Na]+ (calcd for C26H34O11Na+, 545.1993). The 1D NMR data together with HSQC spectrum showed signals for a β-substituted furan ring [δH 7.48 (1H, d, J = 1.5 Hz), 7.42 (1H, dd, J = 1.5, 0.9 Hz) and 6.44 (1H, d, J = 0.9 Hz); δC 144.5, 141.5, 127.9 and 110.0], a β-glucopyranosyl moiety [δH 4.15 (1H, d, J = 7.7 Hz), 3.88 (1H, dd, J = 11.8, 2.3 Hz), 3.64 (1H, dd, J = 11.8, 6.3 Hz), 3.23 (1H, m), 3.20 (1H, m), 3.16 (1H, m) and 3.13 (1H, m); δC 100.6, 78.0, 77.8, 75.3, 72.0 and 63.2], a trisubstituted double bond [δH 6.87 (1H, t, J = 3.6 Hz); δC 138.6 and 135.1] and two methyl groups [δH 1.24 (3H, s) and 1.19 (3H, s); δC 30.0 and 22.2]. The planar structure of 8 was mainly achieved by 1H-1H COSY and HMBC correlations. Analysis of the 1H-1H COSY spectrum revealed key correlations between H-10/H2-1/H2-2/H-3 and H-6/H2-7/H-8, suggesting the presence of two crucial fragments (C-10/C-1/C-2/C-3 and C-6/C-7/C-8). HMBC correlations from H3-19 to C-4, C-5, C-6 and C-10, and from H3-20 to C-8, C-9, C-10 and C-11 aided to establish the 6/6 fused rings of rings A and B. The 1H-1H COSY correlation between H2-11 and H-12, along with HMBC correlations from H-12 to C-13, C-14 and C-16, revealed that the furan ring was attached to C-12. A key HMBC correlation between H-6 and the ester carbonyl at δC 172.3 (C-18) indicated the formation of an intramolecular lactone bridge between C-18 and C-6. Furthermore, a crucial HMBC signal between H-8 and the carboxyl group at δC 177.9 (C-17) defined the attachment position of the carboxyl group. Finally, the HMBC correlation between H-12 (δH 5.25) and the anomeric carbon (δC 100.6) confirmed the linkage of the glucose unit to C-12. The relative configuration of 8 was mainly determined by NOESY correlations and 1H-1H coupling constants. Key NOESY correlations of H3-19/H-6, H-10 and H-6/H-7 at δH 2.27 suggested that these protons were cofacial, leading to a tentative assignment of an α-orientation for these protons. While the NOESY correlations from H3-20 to H-8 and H-7 at δH 1.59, together with the small coupling constants between H-8 and H2-7 (J = 4.4, 3.4 Hz) suggested that these protons were in β-orientation. The coupling constant between H2-11 and H-12, interpreted similarly to the case of 7, suggested an S* configuration at H-12. The absolute configuration of 8 was established as shown by comparing the ECD curve with the calculated one. Thus, compound 8 was assigned as depicted and named tinospinoside M.
The known compounds borapetoside A (9) [21], borapetoside F (10) [22], (2R,5R,6S,9S,10S,12S)-15,16-epoxy-2-hydroxy-6-O-(β-D-glucopyranosyl)-cleroda-3,7,13(16),14-tetraen-17,12-olid-18-oic acid methyl ester (11) [21], tinopanoid T (12) [23], rumphioside Ⅰ (13) [24], borapetoside B (14) [21], borapetoside C (15) [21], tinospinoside C (16) [26], tinosineside A (17) [27], tinosineside B (18) [16], 1-deacetyltinosposide A (19) [27], tinosinenoside B (20) [28] were determined by comparing their NMR data with those in the literature.
2.3 Immunomodulatory activity
Macrophages, a crucial type of immune cell originating from monocytes, are fundamentally important players in the body's immune response. Immune cells secrete cytokines (e.g., NO, TNF-α) to stimulate the immune response, making these molecules useful quantifiers of cellular activation [29, 30]. In current study, all the isolates were first evaluated for their impact on the cell growth of RAW264.7 macrophages but no significant effect was found on cell survival. Further study on the culture supernatant revealed that several diterpenoids could enhance the NO production in RAW264.7 cells, with LPS (200 ng/mL) used as the comparable drug (Table 1). Selected for further study due to its high activity, compound 6 demonstrated the ability to dose-dependently increase the release of NO (Fig. 7A) and TNF-α (Fig. 7B). iNOS, an inducible enzyme that catalyzes NO production from L-arginine, plays a crucial role in the immune system [31], while COX-2 and NO often act synergistically during immune responses [32]. As shown in Fig. 7C, compound 6 effectively upregulated the protein expression of these two enzymes in RAW264.7 cells. NF-κB signaling plays a key role in expressing proteins related to inflammation and immunity, and it has a close relationship with the genes encoding iNOS and COX-2 [33, 34]. Therefore, western blot analysis was employed to determine whether the immunomodulation effect involves the activation of the NF-κB signaling pathway. Results showed that the phosphorylation of NF-κB and IκB-α could be activated by 6 in a dose-dependent manner, while the total amounts of NF-κB and IκB-α were not obviously altered (Fig. 7D). Thus, the immunomodulatory activity of 6 might related to the activation of NF-κB signaling pathway.
The enhancement of NO production of compounds 1 − 20
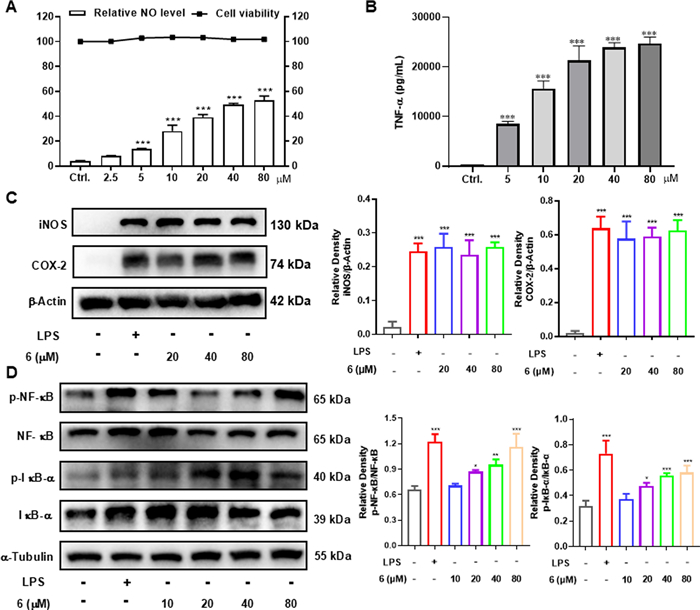
Macrophage immunomodulatory activities of 6. A Relative NO level in the culture supernatant of RAW264.7 cells and cell viability in RAW264.7 cells. B Effect of compound 6 on production of TNF-α in the RAW264.7 cells. C Compound 6 increased the protein levels of iNOS and COX-2 in RAW264.7 cells. D Compound 6 increased the protein levels of p-NF-κB and p-IκB-α in RAW264.7 cells. The data represent the mean ± SD from three independent experiments. *p < 0.05, **p < 0.01 and ***p < 0.001 compared with the control group
3 Experimental
3.1 Plant materials
The tuberous roots of Paratinospora sagittata (Oliv.) Wei Wang were collected in Aug 2018 in Jinghong, Yunnan province, China. A voucher specimen has been deposited at School of Biological Science and Technology, University of Jinan (Accession number: npmc-047).
3.2 Extraction and isolation
The tuberous roots of P. sagittata (30 kg) were air-dried and powdered, then extracted with 95% EtOH at room temperature for three consecutive weeks. The EtOH extract was dissolved in water and partitioned with equal volumes of EtOAc and n-BuOH using a separating funnel. The n-BuOH fraction (760 g) was subjected to chromatography on a D101 macroporous resin column using a gradient of EtOH-water (30%, 50% and 90%) to yield three fractions. The 30% EtOH eluent was further purified by silica gel column chromatography (CC), eluting with a gradient of PE-EtOAc–MeOH (3:1:0 to 0:1:5), resulting in four sub-fractions (A-D). Directed by MS/MS-based molecular networking, diterpenoid-containing fractions B (20 g) and C (56 g) were selected for separation. Fraction B was subjected to silica gel CC and eluted with CH2Cl2- MeOH (100:1 to 5:1) to obtain three subfractions (B1 − B3). Subfraction B2 was separated by RP-C18 CC (MeOH-H2O, 2:8 to 8:2) and Sephadex LH-20 CC (MeOH), yielding B2a−B2c. Subsequent semi-preparative HPLC of B2a (61% MeOH-H2O) gave 15 (2.3 mg, tR = 11 min) and 10 (3.6 mg, tR = 15.5 min); B2b (59% MeOH-H2O) gave 13 (3.7 mg, tR = 18 min), 6 (3.0 mg, tR = 21 min), 5 (8.0 mg, tR = 22.5 min) and 12 (22.3 mg, tR = 29.5 min); B2c (55% MeOH–H₂O) gave 4 (1.2 mg, tR = 12 min), 7 (2.6 mg, tR = 15.5 min) and 3 (3.4 mg, tR = 18 min). Fraction C was subjected to silica gel CC and eluted with PE-EtOAc–MeOH (5:1:0 to 0:1:6) into C1−C3. Subfraction C2 was separated by silica gel CC (CH2Cl2-MeOH, 100:1 to 5:1) to obtain C2a−C2d. C2b was subjected to RP-C18 CC (MeOH-H2O, 3:7 to 7:3), yielding C2b1−C2b4. C2b2 purification via Sephadex LH-20 CC (MeOH) and semi-preparative HPLC (48% MeOH-H2O) afforded 20 (7.8 mg, tR = 13 min), 16 (3.0 mg, tR = 16 min) and 1 (1.5 mg, tR = 18 min). C2b3 was purified by Sephadex LH-20 CC (MeOH), RP-C18 CC (MeOH–H₂O, 3:7 to 10:0), and semi-preparative HPLC (43% MeOH-H2O) to yield 14 (3 mg, tR = 8 min), 11 (4.0 mg, tR = 9.5 min) and 9 (6.0 mg, tR = 10.5 min). C2c was purified by Sephadex LH-20 CC (MeOH) and RP-C18 CC (MeOH-H2O, 3:7 to 10:0), yielding C2c1−C2c5. Semi-preparative HPLC of C2c3 (38% MeCN-H2O) gave 2 (1.7 mg, tR = 10.5 min) and 17 (6.0 mg, tR = 11.5 min), while C2c4 (28% MeCN-H2O) gave 18 (1.5 mg, tR = 15.5 min), 8 (3.6 mg, tR = 18.5 min) and 19 (0.9 mg, tR = 22.5 min).
3.2.1 Tinospinoside F (1)
Colorless gum; [α]D25 − 63 (c 0.34, MeOH); UV (MeOH) λmax (log ε) 211 (3.88); ECD (c 6.5 × 10–4 M, MeOH) λmax (Δε)229 (+ 3.91) nm, 260 (+ 2.30) nm; 1H and 13C NMR data see Tables 2 and 3; HRESIMS (pos.) m/z 543.1836 [M + Na]+ (calcd for C26H32O11Na+, 543.1837).
1H NMR data for compounds 1−8 at 600 MHz
13C NMR data for compounds 1−8 at 150 MHz
3.2.2 Tinospinoside G (2)
Colorless gum; [α]D25 + 26 (c 0.09, MeOH); UV (MeOH) λmax (log ε) 209 (4.14); ECD (c 17 × 10–4 M, MeOH) λmax (Δε) 224 (+ 0.10) nm, 238 (− 0.21) nm, 251 (− 0.08) nm; 1H and 13C NMR data see Tables 2 and 3; HRESIMS (pos.) m/z 556.2393 [M + NH4]+ (calcd for C26H38O12N+, 556.2389).
3.2.3 Tinospinoside H (3)
Colorless gum; [α]D25 + 186 (c 0.20, MeOH); UV (MeOH) λmax (log ε) 207 (3.74); ECD (c 2.6 × 10–4 M, MeOH) λmax (Δε) 210 (+ 3.03) nm, 226 (+ 39.40) nm, 252 (− 10.14) nm; 1H and 13C NMR data see Tables 2 and 3; HRESIMS (pos.) m/z 577.2279 [M + H]+ (calcd for C29H33O12+, 577.2280).
3.2.4 Tinospinoside Ⅰ (4)
Yellow gum; [α]D25 + 122 (c 0.60, MeOH); UV (MeOH) λmax (log ε) 212 (4.25); ECD (c 3.0 × 10–4 M, MeOH) λmax (Δε) 209 (− 3.20) nm, 227 (+ 36.60) nm, 251 (− 11.66) nm; 1H and 13C NMR data see Tables 2 and 3; HRESIMS (pos.) m/z 527.1889 [M + Na]+ (calcd for C26H32O10Na+, 527.1888).
3.2.5 Tinospinoside J (5)
Yellow gum; [α]D25 − 40 (c 0.55, MeOH); UV (MeOH) λmax (log ε) 214 (3.76); ECD (c 4.8 × 10–4 M, MeOH) λmax (Δε) 218 (+ 4.01), 247 (− 3.79) nm; 1H and 13C NMR data see Tables 2 and 3; HRESIMS (pos.) m/z 601.2253 [M + Na]+ (calcd for C29H38O12Na+, 601.2255).
3.2.6 Tinospinoside K (6)
Yellow gum; [α]D25 − 50 (c 0.25, MeOH); UV (MeOH) λmax (log ε) 215 (3.92); ECD (c 3.5 × 10–4 M, MeOH) λmax (Δε) 221 (+ 5.27) nm, 246 (− 5.00) nm; 1H and 13C NMR data see Tables 2 and 3; HRESIMS (pos.) m/z 601.2258 [M + Na]+ (calcd for C29H38O12Na+, 601.2255).
3.2.7 Tinospinoside L (7)
Colorless gum; [α]D25 − 20 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 211 (4.10); ECD (c 0.97 × 10–4 M, MeOH) λmax (Δε) 207 (− 0.67) nm, 222 (+ 6.93) nm, 250(− 5.74) nm; 1H and 13C NMR data see Tables 2 and 3; HRESIMS (pos.) m/z 529.2041 [M + Na]+ (calcd for C26H34O10Na+, 529.2044).
3.2.8 Tinospinoside M (8)
White gum; [α]D25 + 82 (c 0.69, MeOH); UV (MeOH) λmax (log ε) 210 (3.95); ECD (c 3.2 × 10–4 M, MeOH) λmax (Δε) 209 (− 6.71) nm, 252 (+ 3.69) nm; 1H and 13C NMR data see Tables 2 and 3; HRESIMS (pos.) m/z 545.1993 [M + Na]+ (calcd for C26H34O11Na+, 545.1993).
3.3 LC–MS/MS and molecular networking analysis
Details were the same as we formerly described [18].
3.4 Absolute configuration determination of the sugar unit in 1−8
Compounds 1−8 bear a β-glucose/xylose moiety whose absolute configurations were determined by chemical derivatization as we reported before [35], and compounds 1 and 7 were chosen as the model molecule.
3.5 ECD Calculations
The ECD calculations of compounds 1−3 and 8 were performed according to the previously described method [36].
3.6 NO production and cell viability assays
RAW264.7 macrophages (5 × 104 cells) were cultured at 37 ℃ in 96-well flat plates, in the presence or absence of various concentrations of compound 6 for 24 h, in a humidified and 5% CO2-containing incubator. The nitrite accumulation in the culture medium was measured using Griess reagent at 540 nm on the microplate reader. The relative NO level was calculated according to the following formula: [1-(Asample − Ablank)/(ALPS − Ablank)] × 100%. While the viability of the cultured cells was measured by CCK-8 method. 10 μL CCK-8 was added to each well at the final 2–4 h of culture. To the end of the culture, we measured the OD values with microplate reader at 450 nm.
3.7 Measurement of immune cytokine
TNF-α concentrations in culture medium of RAW264.7 macrophages pretreated with presence or absence of various concentrations of compound 6 for 24 h, were measured using commercial ELISA kits following manufacturer protocols. The concentration of TNF-α was calculated from 450 nm OD values using standard curves.
3.8 Western blot analysis
Cells in 6-well plates were treated with varying concentrations of compound 6 for 24 h and LPS (200 ng/mL) is used as a reference standard in current experiment, then lysed in extraction buffer containing protease inhibitors; protein concentrations were determined by BCA assay. Equal protein amounts underwent 10% SDS-PAGE followed by PVDF membrane transfer. After blocking with 5% skim milk, membranes were incubated overnight at 4 ℃ with primary antibodies (iNOS, COX-2, phospho-NF-κB, NF-κB, phospho-IκB-α, IκB-α), washed thrice with TBST, incubated 1 h at room temperature with HRP-conjugated secondary antibody (1:3000), washed again thrice with TBST, and finally detected using ECL on a Chemi-Doc XRS system.
4 Conclusion
Plants of the genus Paratinospora have long been used in Asian traditional medicine to treat immune-related diseases. This study focused on the previously unexplored n-BuOH partition of P. sagittata extract. Guided by MS/MS-based molecular networking, we isolated and identified eight previously undescribed clerodane diterpenoid glycosides and 12 known analogues. Comprehensive spectroscopic analysis elucidated the novel structures, with absolute configurations confirmed by single-crystal X-ray diffraction, TD-DFT/ECD calculations, and chemical derivatization. Selected diterpenoids modulated macrophage activity by enhancing NO production, potentially through NF-κB pathway activation. The current study has confirmed that the clerodane diterpenoids, which are the main chemical constituents of the n-BuOH soluble part of its EtOH extract of P. sagittata, exhibit immunomodulatory activity. These findings demonstrate immunomodulatory potential of P. sagittata, aligning with the traditional uses of Paratinospora species, suggesting that clerodanes may be the pharmacologically active substances responsible for the immune-regulating effects of Paratinospora species. However, due to the limited number of compounds investigated thus far, the structure–activity relationship (SAR) regarding how this structural class promotes macrophage function remains unclear. Further studies with a larger sample size are needed to fully elucidate the SAR.
Notes
Acknowledgements
This project was financially supported by the National Natural Science Foundation of China [No. 82304336], and the Youth Innovation Technology Project of Higher School in Shandong Province [2022KJ096]. The authors were grateful to the authentication of the plant materials by Prof. Guo-hua Ye from Shandong College of Traditional Chinese Medicine.
Author contributions
JSZ: Conceptualization, Funding acquisition, Writing—original draft. RA: Visualization, Investigation. YBP: Investigation. XCZ: Investigation. YKY: Software. JB: Funding acquisition. HZ: Supervision, Writing—review & editing.
Funding
This project was financially supported by the National Natural Science Foundation of China [No. 82304336] and the Youth Innovation Technology Project of Higher School in Shandong Province [2022KJ096].
Availability of data and materials
The data supporting the findings of this study were available on request from the corresponding author, upon reasonable request.
Declarations
Competing interests
The authors declare no competing interest.
References
-
1.Chi SS, She GM, Han D, Wang WH, Liu Z, Liu B. Genus Tinospora: ethnopharmacology, phytochemistry, and pharmacology. Evid-Based Compl Alt. 2016;2016: 9232593. CrossRef PubMed Google Scholar
-
2.Huang MJ, Xiao ZN, Zhang JS, Zhang H. Phytochemistry and pharmacological activities of clerodane diterpenoids from genus Tinospora. Phytochemistry. 2025;237: 114526. CrossRef PubMed Google Scholar
-
3.Huyen PV, Nhung VTT, Phan NHT, Hien NTT, Hanh TTN, Duyen NHH. Phytochemistry and pharmacological activities of Tinospora genus: an updated review. Med Chem Res. 2025;34: 1421-79. CrossRef PubMed Google Scholar
-
4.Wang W, Ortiz RDC, Jacques FMB, Chung SW, Liu Y, Xiang XG, et al. New insights into the phylogeny of Burasaieae (Menispermaceae) with the recognition of a new genus and emphasis on the southern Taiwanese and mainland Chinese disjunction. Mol Phylogenet Evol. 2017;109: 11-20. CrossRef PubMed Google Scholar
-
5.Zhao J, Chen ZH, Huang PC, Chen LW, Zhang MX, Wang LH, et al. Complete mitogenome characteristics and phylogenetic analysis of traditional Chinese medicinal plant Tinospora sagittata (Oliv.) Gagnep. from the Menispermaceae family. Bmc Plant Biol. 2025;25: 165. CrossRef PubMed Google Scholar
-
6.Haque MA, Jantan I, Bukhari SNA. Tinospora species: an overview of their modulating effects on the immune system. J Ethnopharmacol. 2017;207: 67-85. CrossRef PubMed Google Scholar
-
7.Singh B, Nathawat S, Sharma RA. Ethnopharmacological and phytochemical attributes of Indian Tinospora species: a comprehensive review. Arab J Chem. 2021;14: 113381. CrossRef PubMed Google Scholar
-
8.Singh J, Saxena E, Chaudhary AR, Kaur M, Salotra M, Rasane P, et al. Immunomodulatory properties of Giloy (Tinospora cordifolia) leaves and its applications in value-added products. Heliyon. 2025;11: e40948-e40948. CrossRef PubMed Google Scholar
-
9.Editorial Committee of Flora of China, Flora of China (Zhongguo Zhiwu Zhi), Science Press: Beijing; 1996, vol 30 (1), p. 023. PubMed Google Scholar
-
10.State of Administration of Traditional Chinese Medicine of People’s Republic of China. Zhong Hua Ben Cao. Shanghai Scientific and Technical Publishers: Shanghai; 1999, p. 1988−1990. PubMed Google Scholar
-
11.Qin NB, Wang AL, Li DH, Wang KB, Lin B, Li ZL, et al. Cytotoxic clerodane furanoditerpenoids from the root of Tinospora sagittata. Phytochem Lett. 2015;12: 173-6. CrossRef PubMed Google Scholar
-
12.Li G, Ding W, Wan F, Li Y. Two New Clerodane Diterpenes from Tinospora sagittata. Molecules. 2016;21: 1250. CrossRef PubMed Google Scholar
-
13.Zhang G, Ma H, Hu S, Xu H, Yang B, Yang Q, et al. Clerodane-type diterpenoids from tuberous roots of Tinospora sagittata (Oliv.) Gagnep. Fitoterapia. 2016;110: 59-65. CrossRef PubMed Google Scholar
-
14.Li W, Huang C, Liu Q, Koike K. Bistinospinosides A and B, dimeric clerodane diterpene glycosides from Tinospora sagittata. J Nat Prod. 2017;80: 2478-83. CrossRef PubMed Google Scholar
-
15.Song J-Q, Deng L, Zhu Y-L, Zhou H, Kong XQ, Zhang LJ, et al. Clerodane diterpenoids with in-vitro anti-neuroinflammatory activity from the tuberous root of Tinospora sagittata (Menispermaceae). Phytochemistry. 2024;218: 113932. CrossRef PubMed Google Scholar
-
16.Zhang JS, Hu YT, Song KS, Wu F, Zhu KK, Xu DF, et al. Diterpenoid glucosides with cystathionine γ-lyase inhibitory activity from Tinospora sinensis. Bioorg Chem. 2021;116: 105400. CrossRef PubMed Google Scholar
-
17.Zhang JS, Xu DF, Wang YY, Ma RF, Zhang H. Clerodane furanoditerpenoids from the stems of Tinospora sinensis. Arch Pharm Res. 2022;45: 328-39. CrossRef PubMed Google Scholar
-
18.Ao R, Li MY, Yang FF, Bao J, Zhang JS, Zhang H. Targeted discovery of clerodane diterpenoids from Tinospora sinensis as immunomodulatory agents. Fitoterapia. 2024;178: 106174. CrossRef PubMed Google Scholar
-
19.Huang C, Li W, Ma F, Li Q, Asada Y, Koike K. Tinospinosides D, E, and Tinospin E, further clerodane diterpenoids from Tinospora sagittata. Chem Pharm Bull. 2012;60: 1324-8. CrossRef PubMed Google Scholar
-
20.Li W, Huang C, Li S, Ma F, Li Q, Asada Y, et al. Clerodane diterpenoids from Tinospora sagittata (Oliv.) Gagnep. Planta Med. 2012;78: 82-5. CrossRef PubMed Google Scholar
-
21.Choudhary MI, Ismail M, Shaari K, Abbaskhan A, Sattar SA, Lajis NH, et al. cis-clerodane-type furanoditerpenoids from Tinospora crispa. J Nat Prod. 2010;73: 541-7. CrossRef PubMed Google Scholar
-
22.Phan VK, Chau VM, Nguyen TD, La VK, Dan TH, Nguyen HN, et al. Aporphine alkaloids, clerodane diterpenes, and other constituents from Tinospora cordifolia. Fitoterapia. 2010;81: 485-9. CrossRef PubMed Google Scholar
-
23.Zhu YL, Deng L, Dai XY, Song JQ, Zhu Y, Liu T, et al. Tinopanoids K-T, clerodane diterpenoids with anti-inflammatory activity from Tinospora crispa. Bioorg Chem. 2023;140: 106812. CrossRef PubMed Google Scholar
-
24.Martin TS, Ohtani K, Kasai R, Yamasaki K. Furanoid diterpene glucosides from Tinospora rumphii. Phytochemistry. 1996;42: 153-8. CrossRef PubMed Google Scholar
-
25.Gao Y, Niu Y-F, Wang F, Hai P, Wang F, Fang Y-D, et al. Clerodane diterpenoids with anti-hyperglycemic activity from Tinospora crispa. Nat prod Bioprospect. 2016;6: 247-55. CrossRef PubMed Google Scholar
-
26.Kabbashi AS, Sattar MA, Aamer M, Siddiqui NN, Kamran M, Fayaz A, et al. Clerodane furanoditerpenoids from Tinospora bakis (A.Rich.) Miers (Menispermaceae). Molecules. 2024;29: 154. CrossRef PubMed Google Scholar
-
27.Dong L-P, Chen C-X, Ni W, Xie B-B, Li J-Z, Liu H-Y. A new dinorclerone diterpenoid glycoside from Tinospora sinensis. Nat Prod Res. 2010;24: 13-7. CrossRef PubMed Google Scholar
-
28.Jiang H, Zhang G-J, Liu Y-F, Wang H-S, Liang D. Clerodane diterpenoid glucosides from the stems of Tinospora sinensis. J Nat Prod. 2017;80: 975-82. CrossRef PubMed Google Scholar
-
29.Chen SZ, Saeed AFUH, Liu Q, Jiang Q, Xu HZ, Xiao GG, et al. Macrophages in immunoregulation and therapeutics. Signal Transduct Target Ther. 2023;8: 207. CrossRef PubMed Google Scholar
-
30.Lundberg JO, Gladwin MT, Weitzberg E. Strategies to increase nitric oxide signalling in cardiovascular disease. Nat Rev Drug Discov. 2015;14: 623-41. CrossRef PubMed Google Scholar
-
31.Kashfi K, Kannikal J, Nath N. Macrophage reprogramming and cancer therapeutics: role of iNOS-derived NO. Cells-Basel. 2021;10: 3194. CrossRef PubMed Google Scholar
-
32.Murakami A, Ohigashi H. Targeting NOX, INOS and COX-2 in inflammatory cells: chemoprevention using food phytochemicals. Int J Cancer. 2007;121: 2357-63. CrossRef PubMed Google Scholar
-
33.Capece D, Verzella D, Flati I, Arboretto P, Cornice J, Franzoso G. NF-κB: blending metabolism, immunity, and inflammation. Trends Immunol. 2022;43: 757-75. CrossRef PubMed Google Scholar
-
34.Surh YJ, Chun KS, Cha HH, Han SS, Keum YS, Park KK, et al. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: down-regulation of COX-2 and iNOS through suppression of NF-kappa B activation. Mutat Res. 2001;480–481: 243-68. PubMed Google Scholar
-
35.Ao R, Li M-H, Xiao Z-N, Tang X-L, Zhang J-S, Zhang H. Sesquiterpenes from Tinospora sinensis and their chemotaxonomic significance. Biochem Syst Ecol. 2024;116: 104877. CrossRef PubMed Google Scholar
-
36.Zhang JS, Xu DF, Cao XX, Wang YY, Zhang H. Lignans with NO inhibitory activity from Tinospora sinensis. Chin J Nat Medicines. 2021;19: 500-4. CrossRef PubMed Google Scholar
Copyright information
© The Author(s) 2026
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
