
Harziachalasins A–G, polycyclic-fused cytochalasins from the endophytic fungus Trichoderma harzianum MLJ-4 with HIV latency reversal activity

Abstract
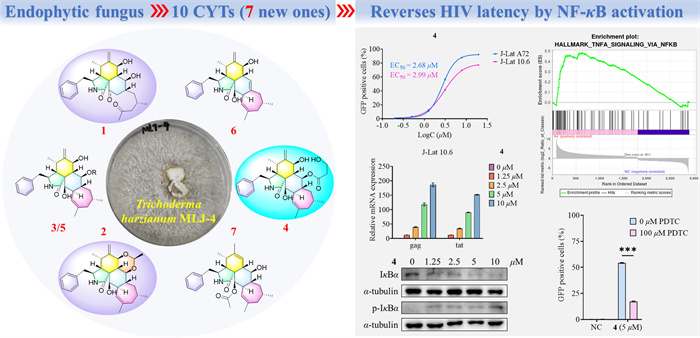
Seven new polycyclic-fused cytochalasins (CYTs), harziachalasins A–G (1–7), together with three known analogues (8–10) were isolated from the solid culture of the endophytic fungus Trichoderma harzianum MLJ-4, which was originally isolated from the leaves of Asclepias curassavica. The planar and absolute structures of all new compounds were determined on the basis of extensive spectroscopic data (1D, 2D NMR and HR-ESI–MS), NMR calculations with DP4+probability analysis, and theoretical simulations of ECD spectra. Compound 1 represents the first example of 5/6/6 tricyclic CYT featuring a 2-methyl-4-oxopentyl side chain at the C-14 position. This novel architecture originates from a 5/6/6/7 tetracyclic CYT precursor through sequential oxidative cleavage of the C-19–C-20 bond followed by decarboxylative elimination of C-19. Compound 2 features an unprecedented 5/6/6/6/7 pentacyclic scaffold incorporating a 1,3-dioxane moiety, may be constructed by the acetalization of the 7-OH and 13-OH on a 5/6/6/7 tetracyclic CYT with acetaldehyde. Compounds 1–10 were screened for HIV latency reversal activity using J-Lat A72 and J-Lat 10.6 cell models. Compound 4 showed strong activity, with half-maximal effective concentrations (EC50) values of 2.68 μM (J-Lat A72 cells) and 2.99 μM (J-Lat 10.6 cells), demonstrating consistent potency. Mechanistic studies revealed 4 activated the NF-κB pathway to reverse HIV latency, offering insights for new therapeutic strategies targeting this pathway.Graphical Abstract
Keywords
Endophytic fungus Trichoderma harzianum Polycyclic-fused cytochalasins HIV latency reversal activity1 Introduction
Fungi represent an underexploited reservoir of structurally diverse natural products with significant bioactivities, serving as invaluable sources for both therapeutic development and molecular probes for target discovery [1]. Among these metabolites, cytochalasins (CYTs) constitute a distinctive class of fungal-derived compounds biosynthesized via polyketide synthase-nonribosomal peptide synthetase (PKS-NRPS) pathways, integrating amino acids and polyketide precursors [2, 3]. Characterized by an isoindole moiety fused with a macrocyclic ring system, their structural complexity arises from: (1) Varied amino acid incorporation, (2) PKS-NRPS-mediated chains modifications, and (3) intramolecular cyclization forming polycyclic systems [4, 5]. Recent advances include the discovery of novel scaffolds [6–17], mechanistic studies [18, 19], and synthetic breakthroughs [5, 20–22], highlighting their pharmaceutical potential.
Despite antiretroviral therapy (ART) effectively suppressing HIV replication, persistent viral reservoirs remain the primary barrier to a cure [23, 24]. Latency-reversing agents (LRAs) have emerged as a strategic therapeutic approach to activate latent HIV-1, enabling immune clearance [25–27]. Notably, naturally occurring CYTs−highly oxygenated polycyclic metabolites−demonstrate compelling anti-HIV activities, positioning them as promising LRA candidates [28, 29]. Building upon our ongoing exploration of fungal-derived bioactive natural products [30–37], the remarkable structural diversity and therapeutic potential of CYTs emerged as a key research focus in our group. In particular, the strain Trichoderma harzianum MLJ-4 was selected for detailed investigation based on its promising metabolic profile. This study led to the isolation of seven new CYTs, harziachalasins A–F (1–7) (Fig. 1), along with three known analogs (8–10) (Fig. 1), phomopsischalin B (8) [6], phomopsischalin A (9) [6], and ueckerchalasin A (10) [38]. Herein, we describe their isolation, structural elucidation, and evaluation of HIV latency reversal activity.
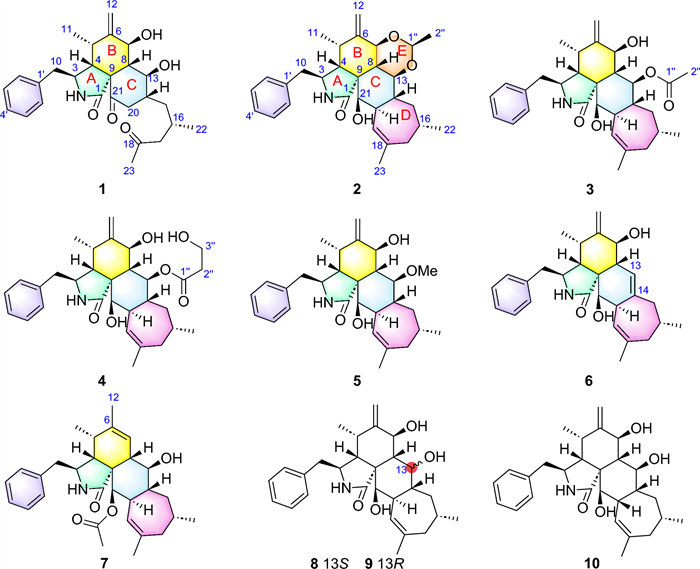
The structures of polycyclic-fused cytochalasins (CYTs)
2 Results and discussion
2.1 Structure elucidation
Harziachalasin A (1) was obtained as white, amorphous powder. Its molecular formula C27H35NO5 was determined by the HR-ESI-MS ion at m/z 476.2384 [M+Na]+ (calcd. 476.2407), requiring 11 degrees of unsaturation. The IR absorption bands revealed the presence of hydroxyl (3330 cm−1), carbonyl (1676 cm−1), and phenyl (1600 and 1456 cm−1) functionals. The 1H NMR spectrum recorded in CDCl3 (Table 1) displayed one mono-substituted benzene ring in the aromatic region [δH 7.19 (2H, d, J=7.3 Hz), 7.24 (1H, t, J=7.3 Hz), and 7.31 (2H, t, J=7.3 Hz)], an terminal methylene [δH 5.01 (1H, s) and 5.20 (1H, s)], two sp3 oxygenated methines [δH 4.46 (1H, d, J=9.2 Hz) and 4.98 (1H, td, J=9.9, 1.8 Hz)], three sp3 methyl groups [δH 0.89 (3H, d, J=6.6 Hz), 0.93 (3H, d, J=6.6 Hz), and 2.15 (3H, s)], and other aliphatic protons. The 13C and DEPT NMR experiments indicated 27 carbons including two ketocarbonyls [δC 209.7 (C) and 203.9 (C)], an amide carbonyl [δC 172.1 (C)], a mono-substituted benzene ring [δC 126.9 (CH), 128.8 (CH×2), 129.3 (CH×2), and 137.6 (C)], one exocyclic double bond [δC 113.6 (CH2) and 148.5 (C)], eight sp3 methines [including two oxygenated ones at δC 73.9 (CH) and 72.5 (CH)], three sp3 methyls [δC 12.6 (CH3), 20.3 (CH3), and 30.5 (CH3)], a sp3quaternary carbon [δC 62.3 (C)], and four sp3 methylenes. As eight of 11 degrees of unsaturation were occupied by two carbonyls, an amide carbonyl, a benzene ring, and a double bond, the remaining three degrees of unsaturation indicated that 1 was a tricyclic skeleton. The above-mentioned information together with the comparison of its NMR data with the co-isolated known compounds 8–10 [6, 38] suggested that 1 should be a tricyclic ring system derived from the type of 5/6/6/7-tetracyclic cytochalasin.
1H (400 MHz) and 13C (100 MHz) NMR Data of Compounds 1 and 2 in CDCl3 (δ in ppm)
The planar structure of 1 was established by extensive analysis of 2D NMR spectra. As shown in Fig. 2, the observed two 1H–1H COSY spin systems: H-10/H-3/H-4/H-5/H-11 and H-7/H-8/H-13/H-14(/H-20)/H-15/H-16/H-17, suggested the present of two key fragments, a (C-10–C-3–C-4–C-5–C-11) and b (C-7–C-8–C-13–C-14(–C-20)–C-15–C-16–C-17). By analysis of the HMBC correlations (Fig. 2), from 2-NH to C-4 and C-9, H-4 to C-6 and C-1, H-7 to C-5 and C-13, H-8 to C-1, C-4, C-9, C-14, and C-21, H2-12 to C-5, C-6, and C-7, proved that fragment a, the exocyclic double bond, part of moiety b, one ketocarbonyl, the only sp3 quaternary carbon, and the amide carbonyl group construct a 5/6/6 tricyclic core. The HMBC correlations from H3-23 to another ketocarbonyl (C-18) and C-17 indicated that fragment b ended with an acetyl group. Thus, a 2-methyl-4-oxopentyl side chain was determined to be connected to ring C, rather than being fused with a 7-membered ring as in the skeleton of 5/6/6/7-tetracyclic cytochalasin. Furthermore, key HMBC correlations from H-2′/H-6′ to C-10 and form H-10 to C-1′ confirmed the connection of the mono-substituted benzene ring to ring A. Here, the planar structure of 1 was elucidated.
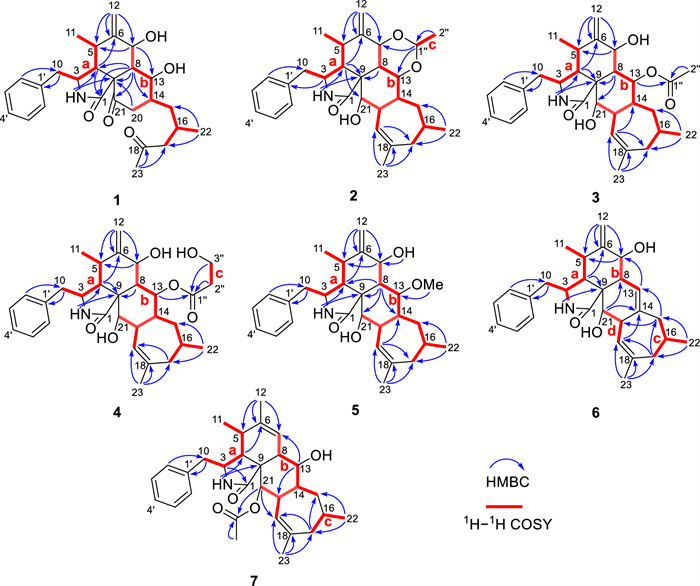
1H−1H COSY and key HMBC correlations of 1−7
The relative configurations of C-3, C-4, C-5, C-7, C-8, and C-9 in 1 were established to be the same as those of phomopsischalin B (8) [6] by comparison of their 1D NMR and NOESY data. In particular, H-8 exhibited a typical triple peak with a coupling constant of 9.6 Hz, indicating that H-7 and H-8 together with H-13 and H-8 were both in trans configuration. Therefore, it was arbitrarily designated that H-8 was β-oriented, while H-7 and H-13 were α-oriented. The NOE interactions of H-5/H-8, H-4/H-8, and H-14/H-8 indicated that all these protons were β-orientation (Fig. 3), and the observed NOE correlation of H-3/H3-11 suggested the α-orientation of H-3. According to the biosynthetic pathway, the relative configuration of H-16 was determined to be the same as the β-orientation of 8. The absolute configuration of 1 was assigned by comparison of its experimental ECD spectrum with those calculated for two presumable isomers based on time-dependent density functional theory (TDDFT) (Fig. 4A).
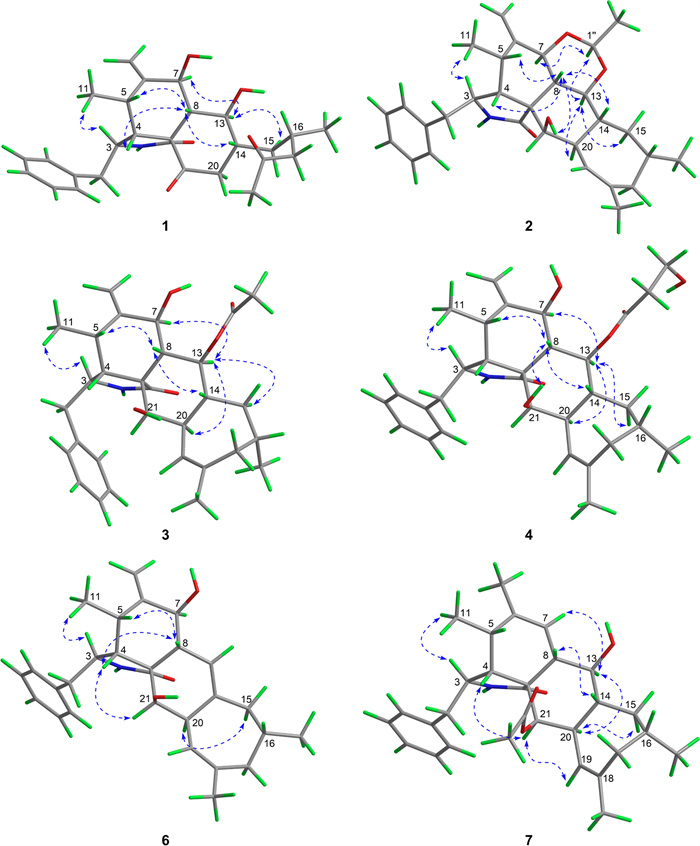
Key NOE correlations of 1−4 and 6−7
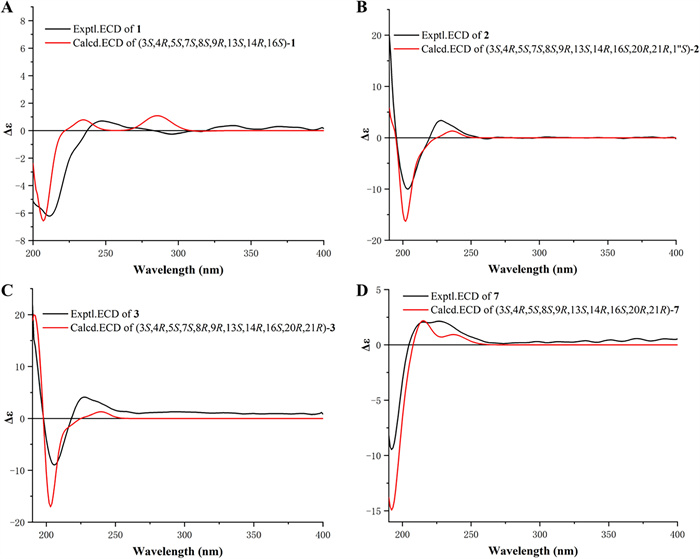
Experimental and calculated ECD curves (A−C) of 1−3 and 7 (D)
Therefore, the structure of compound 1 was identified as an unprecedented 19,20-seco-19-nor-5/6/6/7-fused tetracyclic cytochalasin scaffold. It represents the first example of 5/6/6-fused tricyclic cytochalasin featuring a 2-methyl-4-oxopentyl side chain, which is likely derived from phomopsischalin B (8) [6] by oxidative ring-opening and decarboxylation (Scheme 1).
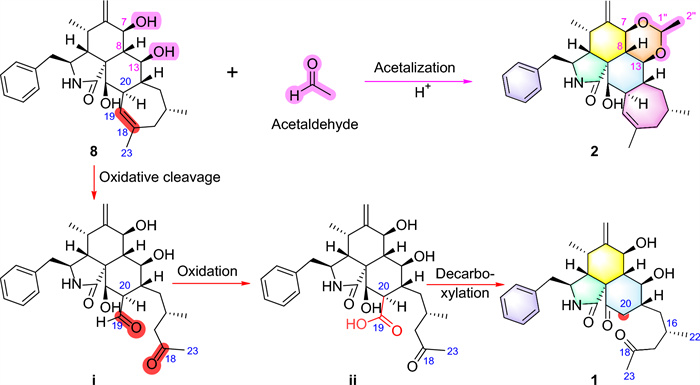
Proposed Biosynthetic Pathways for 1 and 2
Harziachalasin B (2) exhibited a molecular formular of C30H39NO4 with 12 degrees of unsaturation based on the HR-ESI-MS data (m/z 500.2776 [M + Na]+, calcd. 500.2771). Its spectroscopic data (Table 1) bore a resemblance to those of 8, except for the presence of an additional ethane-1,1-dioxy group [δH 1.36 (3H, d, J=5.1 Hz) and 4.82 (1H, q, J=5.1 Hz); δC 21.3 (CH3) and 99.4 (CH)], along with the changes in the chemical shifts of the oxygenated methines C-7 (δC 75.4) and C-13 (δC 78.1) and their adjacent carbons in 2. This information suggested that the newly emerged C2 unit may be associated with C-7 or/and C-13. Combined with an additional degree of unsaturation (12 degrees of unsaturation for 2; 11 degrees of unsaturation for 8) and the HMBC correlations from H-7 and H-13 to C-1′′ (δC 99.4) and from H-1′′ to C-7 and C-13, it can be determined that the newly introduced ethane-1,1-dioxy group formed a new 1,3-dioxane ring with the B/C rings. Therefore, a 5/6/6/6/7-fused pentacyclic ring system skeleton for 2 was constructed. The relative configuration of the newly formed chiral center (C-1′′) was determined as S* by the observed NOE correlations of H-7/H-1′′ and H-13/H-1′′. In addition, no correlations between H-16 and other hydrogen atoms were observed in the NOESY spectrum. This absence of signals may be attributed to significant overlap in the high-field region, which could obscure the relevant cross-peaks. Ultimately, the configuration of H-16 was unambiguously assigned as 16S through NMR chemical shift calculations combined with DP4 + probability analysis. Between the two possible stereoisomers, 16S-2a and 16R-2b, only the 16S configuration yielded a DP4 + probability greater than 100.00% (Figure S2), thereby confirming its absolute configuration. The relative configurations of other chiral centers were confirmed to be the same as 8 by 1D NMR and NOE correlations (Fig. 3) analysis. Considering the strong resemblance between the experimental and calculated ECD curves (Fig. 4B), the absolute configuration of 2 was thereby assigned to be 3S,4R,5S,7S,8S,9R,13S,14R,16S,20R,21R,1′′S. Thus, the structure of 2 was determined as shown, which represents the first example of 5/6/6/6/7-fused pentacyclic ring system skeleton for cytochalasins. The plausible biosynthetic pathway of 2 may be the condensation of the 7-OH and 13-OH on compound 8 with acetaldehyde (Scheme 1).
Harziachalasin C (3) possessed the molecular formular C30H39NO5 with 12 degrees of unsaturation, by HR-ESI–MS ([M + Na]+ m/z 516.2712, calcd. 516.2720). Interpretation of NMR data (Table 2) established the carbocyclic skeleton of 3 being identical with that of 8, except for the presence of an additional acetoxy group [δH 2.07 (3H, s); δC 21.9 (CH3) and 171.8 (C)] in 3. By analysis of the HMBC correlations (Fig. 2) from H-13 [δH 5.80 (1H, t, J=10.2 Hz)] to C-1′′ (δC 171.8), it confirmed the attachment of the acetoxy group at C-13. The relative configuration of 3 was consistent with that of 8 according to comparable observations in the 1D NMR and NOESY experiments (Fig. 3). The identical experimental and calculated ECD curves (Fig. 4C) ascertained its absolute configuration as shown.
1H (400 MHz) and 13C (100 MHz) NMR Data of Compounds 3 and 4 in CDCl3 (δ in ppm)
Harziachalasin D (4) possessed the molecular formula of C31H41NO6 as established from the ion peak at m/z 524.2988 [M + H]+ (calcd. 524.3007) in its HR-ESI-MS spectrum. Comparison of the spectroscopic data (Table 2) implied that 4 possessed a 3-hydroxypropionyloxy group [δH 2.98 (2H, m) and 4.66 (2H, t, J=6.3 Hz); δC 32.0 (CH2), 70.2 (CH2), and 170.0 (C)] instead of the acetoxy in 3, which could be further confirmed by the 1H–1H COSY correlations of H-2′′/H-3′′ and the HMBC correlations from H-2′′ and H-3′′ to C-1′′ (Fig. 2). The location of the 3-hydroxypropionyloxy group at C-13 was supported by the HMBC correlation from H-13 to C-1′′. The analysis of the NOESY correlations (Figure S1) and the experimental ECD spectrum (Fig. 5) confirmed that the stereochemistry of 4 was identical to that of 3.
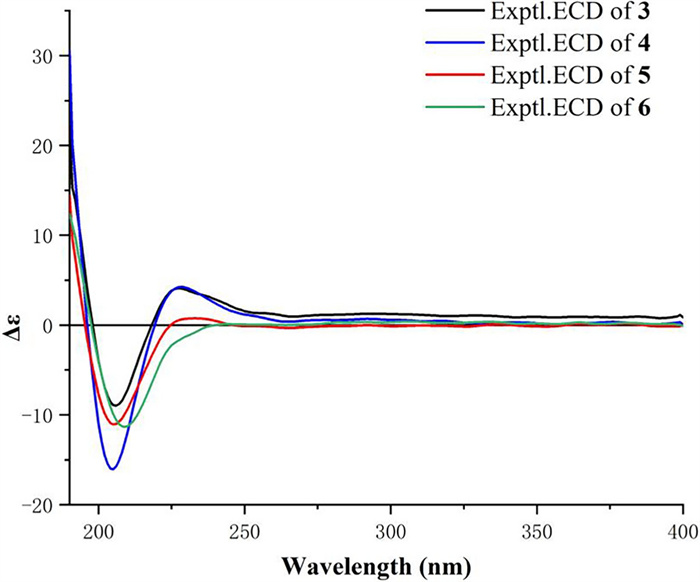
Experimental ECD curves of 3−6
Harziachalasin E (5) displayed a sodium adduct HR-ESI-MS ion at [M + Na]+ m/z 488.2764 (calcd. 488.2771), indicating the molecular formula C29H39NO4 with 11 degrees of unsaturation. Its spectroscopic data (Table 3) bore a close resemblance to those of 3, except for the appearance of a methoxy (δH 3.55; δC 60.1) rather than the acetoxy in 3. The location of the methoxy at C-13 was verified by the important HMBC correlation from the protons of the methoxy to the oxygenated methine [δC 85.6 (CH, C-13)] (Fig. 2). By analyzing its 1D NMR data and NOE correlations, it can be determined that the relative configurations of compounds 5 and 3 were the same (Figure S1). The similar ECD curves of compounds 3−5 (Fig. 5) suggested these compounds shared the same absolute configurations.
1H (400 MHz) and 13C (100 MHz) NMR Data of Compounds 5−7 in CDCl3 (δ in ppm)
Harziachalasin F (6) had the molecular formula C28H35NO3 as derived from HR-ESI-MS analysis (m/z 434.2681 [M + H]+, calcd. 434.2690). Its NMR data (Table 3) for 6 were also very similar to those of 8, with the apparent differences being due to the presence of an additional trisubstituted double bond [δH 5.64; δC 118.7 (CH) and 136.8 (C)] instead of two methines including an oxygenated one, which suggested that 6 was a dehydration product of 8. Detailed analysis of their 2D NMR data confirmed the above deduction, especially by the observation of HMBC correlations from H-7 to C-13 and from H2-15 to C-13 (Fig. 2). The relative configurations of C-3, C-4, C-5, C-7, C-8, C-9, C-16, and C-20 in 6 were determined to be the same as 8 by comparing their 1D NMR data and the observed NOE correlations (Fig. 3). The experimental ECD spectrum (Fig. 5) showed the absolute configuration of 6 was identical to that of 3−5.
Harziachalasin G (7) possessed a molecular formula of C30H39NO4, as determined by the HR-ESI-MS ion at m/z 478.2943 [M + H]+ (calcd. for 478.2952). The 1D NMR data (Table 3) of 7 exhibited similarities to those of 3, except for the disappearance of a terminal methylene and an oxygenated methine in 3 instead of the presence of a singlet methyl and an olefinic methine, which indicated that the Δ6(12) double bond in 3 was migrated to Δ6 in 7. This hypothesis was further confirmed by the 1H−1H COSY of H-8/H-7 and the HMBC correlations from H3-12 to C-5, C-6, and C-7. However, the acetoxy was located at C-21 instead of at C-13, which was supported by the HMBC correlation from H-21 to the ester carbonyl (δC 171.4) (Fig. 2). The relative configuration of 7 was assigned as shown by analysis of the NOESY spectrum (Fig. 3). Finally, the (3S,4R,5S,8S,9R,13S,14R,16S,20R,21R) absolute configuration of 7 was elucidated by the ECD calculation (Fig. 4D).
Three known analogues, phomopsischalin B (8) [6], phomopsischalin A (9) [6], and ueckerchalasin A (10) [38] were identified by comparison of their experimental spectroscopic data with those reported.
2.2 Screening for HIV latency reversing agents (LRAs)
The ten isolated natural CYTs (1−10) were evaluated for their ability to reverse HIV latency using two well-established cell models: J-Lat A72 and J-Lat 10.6 [39], and wikstroelide E served as the reference compound in the experimental group [40]. Following a 24 h incubation period with each compound at a concentration of 10 μM, flow cytometry analysis demonstrated that 4 exhibited the most potent latency-reversing activity in both cell lines (Fig. 6A). Dose–response studies revealed that 4 displayed significant potency, with half-maximal effective concentration (EC50) of 2.68 μM in J-Lat A72 cells and 2.99 μM in J-Lat 10.6 cells (Fig. 6B). Notably, the compound exhibited no cytotoxic effects on normal cells, as confirmed by cell viability assays (Fig. S3), with an IC50 value exceeding 40 μM in HEK-293 T cells. Representative flow cytometry scatter plots (Fig. 6C) and corresponding fluorescence microscopy images (Fig. 6D) confirmed the dose-dependent nature of HIV reactivation by 4. Beyond GFP expression, quantitative RT-qPCR analysis demonstrated that 4 also induced dose-dependent upregulation of key HIV transcripts, including gag and tat mRNA (Fig. 6E). This multi-parameter validation strengthens the conclusion that compound 4 represents a promising lead for HIV latency reversal.
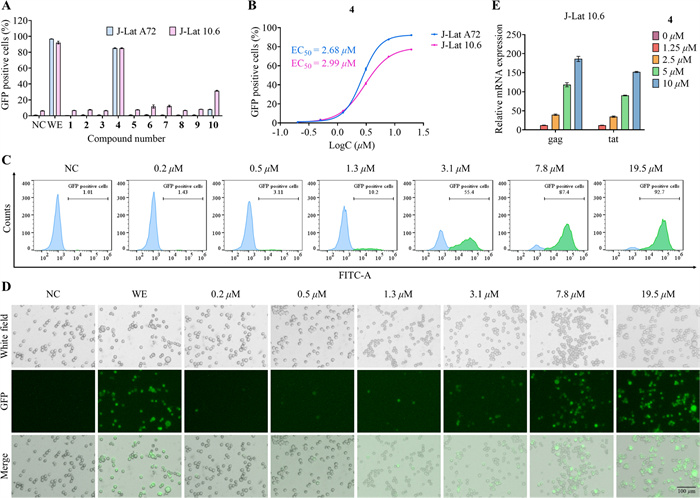
Evaluation of HIV latency reversing agent activity of CYTs 1−10. A Flow cytometry analysis for screening HIV latency reversing agents. B EC50 curves of 4 in J-Lat A72 and J-Lat 10.6 cell lines. C Representative flow cytometry scatter plots demonstrating the HIV latency reversal effect of 4 at various concentrations. D Representative fluorescence microscopy images illustrating the HIV latency reversal effect of 4 at various concentrations. E Relative expression levels of HIV gag and tat mRNAs in J-Lat 10.6 cells treated with 4 at different doses for 24 h. Negative control (NC): Cells were treated with vehicle (DMSO). Wikstroelide E (WE): Positive control, cells were treated with 50 nM WE
2.3 Compound 4 reverses HIV latency via activating the NF-κB pathway, and preliminary RNA-seq analysis suggests potential dual antiviral-antitumor effects
To elucidate the mechanism of compound 4 as an HIV LRA, we conducted RNA sequencing in the J-Lat 10.6 cell line following 24 h treatment with either DMSO (negative control, NC) or 10 μM compound 4 (sample name: A4). Comparative analysis revealed 3464 significantly upregulated genes and 1750 markedly downregulated genes (Fig. 6A). Gene set enrichment analysis (GSEA) further demonstrated that compound 4 strongly activates the NF-κB signaling pathway (Fig. 7B and C).
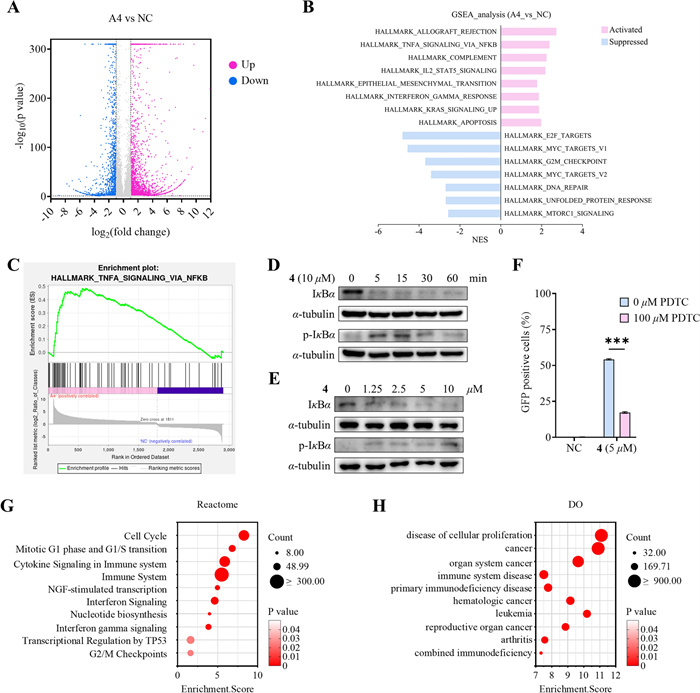
Compound 4 reprograms transcriptomes in the HIV latent cell model with experimental validation of NF-κB pathway activation. A Volcano plot shows significantly different expressed genes in J-Lat 10.6 cells treated with or without 10 μM of 4 for 24 h. B GSEA enrichment result of the Hallmark gene set. C Positive enrichment of the Hallmark TNF-α signaling via NF-κB gene set in compound 4-treated cells. D IκBα degradation and phosphorylation induced by 10 μM compound 4. E Dose-dependent IκBα degradation and phosphorylation induced by 4 after 15-min treatment. F PDTC attenuates the HIV latency reversal effect of 4. G Bubble plot depicting enriched Reactome pathways results. H Results from Disease Ontology (DO) enrichment analysis were demonstrated by bubble plot
Previous studies have established that latent HIV-1 reactivation is mediated by the NF-κB signaling pathway [41–43]. To systematically evaluate whether the latency-reversing activity of compound 4 operates through this canonical pathway, we conducted a series of mechanistic investigations focusing on key regulatory components of NF-κB activation. Western blot analysis was employed to quantify the protein levels of IκBα and its phosphorylated form (p-IκBα), both are critical regulators in the canonical NF-κB activation cascade. Time-course (0−60 min) and dose–response (0−10 μM) experiments were performed to characterize the dynamic effects of 4 on these molecular targets. As shown in Fig. 7D and E, compound 4 promoted the phosphorylation of IκBα in a concentration-dependent manner, as reflected by the initial increase in p-IκBα levels. This was followed by a subsequent degradation of p-IκBα, resulting in an overall reduction in both p-IκBα and IκBα protein levels over time. To confirm the pathway specificity, we employed ammonium pyrrolidine dithiocarbamate (PDTC), a well-characterized inhibitor of NF-κB signaling pathway. Pretreatment with 100 μM PDTC significantly attenuated the latency-reversing capacity of compound 4 by approximately 69% (Fig. 7F), providing strong pharmacological evidence for NF-κB pathway involvement. Collectively, these data establish that compound 4 mediates HIV-1 latency reversal primarily through activating canonical NF-κB pathway. This mechanistic understanding positions 4 as a valuable chemical tool for further investigation of NF-κB-dependent latency reversal strategies.
Our integrated RNA-seq and experimental validation demonstrate that compound 4 exerts its HIV latency-reversing activity by NF-κB pathway activation, a mechanism consistent with established HIV-1 reactivation models. Transcriptomic profiling further revealed its modulation of cancer-associated pathways, including cytokine signaling, cell cycle, immune responses, and DNA repair (Fig. 7B and G), consistent with the anticancer effects of related CYTs (e.g., cell cycle arrest) [44–46]. These multifaceted effects collectively explain its association with immune-related diseases and multiple cancers (Fig. 7H). This dual functionality addresses a critical unmet need: HIV patients exhibit higher cancer incidence, with cancer accounting for 20% of deaths despite viral suppression [47–49]. We thus propose a novel therapeutic strategy−NF-κB-involved HIV reservoir clearance combined with oncogenic pathway modulation−which is especially relevant given HIV patients’ elevated cancer risk. To assess compound 4’s broader therapeutic potential, in vitro and in vivo anti-tumor efficacy studies will be conducted to validate these findings.
3 Conclusions
Chemical investigation of the endophytic fungus T. harzianum MLJ-4 isolated from the medicinal plant A. curassavica led to the discovery of ten CYTs (1−10) from its rice fermentation extract, including seven novel metabolites designated harziachalasins A−G (1−7) characterized through comprehensive spectroscopic analysis. This study unveiled two unprecedented structural frameworks in CYTs chemistry: compound 1 represents the first 19,20-seco-19-nor-5/6/6-fused tricyclic scaffold with a distinctive 2-methyl-4-oxopentyl side chain at C-14, establishing a novel subclass, while compound 2 features an exceptional 1,3-dioxane-incorporated E-ring forming the inaugural 5/6/6/6/7-fused pentacyclic system in this class. These findings significantly expand the structural diversity of the rare 5/6/6/7-tetracyclic CYTs, with only seven prior analogues reported including ueckerchalasin C [38], diaporchalasin D [50], diaporchalasin E [50], and curtachalasin P [51] together with compounds 8−10 isolated by this work. Biological evaluation demonstrated that compound 4 effectively reverses HIV latency (EC50 = 2.68 μM in J-Lat A72; 2.99 μM in J-Lat 10.6 cells) by activating the NF-κB pathway. This mechanism was confirmed through three lines of evidence: (1) RNA-seq analysis showing NF-κB pathway upregulation, (2) IκBα degradation/phosphorylation, and (3) significant attenuation of activity by the NF-κB inhibitor PDTC. Notably, RNA-seq also revealed an additional therapeutic benefit: compound 4 modulated cancer-related pathways, potentially reducing the elevated cancer risk observed in HIV patients. The structural innovations coupled with mechanistically elucidated bioactivity highlight both the metabolic versatility of Asclepias-associated endophytes and the potential of these derivatives as valuable leads for combined HIV/cancer therapy, positioning them as promising candidates for further structure–activity optimization and in vivo efficacy studies.
4 Experimental section
4.1 General experimental procedures
MCP 200 modular circular polarimeter from Anton Paar was used to determine the optical rotations. An Applied Photophysics Chirascan spectrometer was used to measure ECD and UV data. A PerkinElmer Spectrum Two FTIR spectrometer with a UATR accessory was applied to collect IR spectra. 1D and 2D NMR spectra were recorded using Bruker Avance Ⅲ 400 and Bruker Ascend TM 500 spectrometers. A Micromass Q-TOF spectrometer from Waters was used to receive HR-ESI–MS data. Column chromatography (CC) fractionations mainly relied silica gel (100–200, 200–300, and 300–400 mesh, Qingdao Haiyang Chemical Co., Ltd.), reversed-phase C18 (Rp-C18) silica gel (12 nm, S-50 μm, YMC Co., Ltd.), and Sephadex LH-20 gel (Amersham Biosciences). High-performance liquid chromatography (HPLC) separations were conducted on a Shimadzu LC-20 AT with an SPD-M20A PDA detector (Kyoto, Japan) using a YMC-pack ODS-A column (10 × 250 mm, S-5 μm) and a NanoChrom ChromCoreTM 5–120 C18 column (250 × 10 mm, 5 µm). Chemical solvents were of analytical grade (Guangzhou Chemical Reagents Company, Ltd.) while acetonitrile (MeCN) was of HPLC grade (Grace Chemical Technology Co., Ltd.).
4.2 Fungal materials
The strain T. harzianum MLJ-4 was isolated from the plant A. curassavica, which was collected from South China Botanical Garden of Guangzhou city. The strain was identified by sequence analysis of rDNA ITS (internal transcribed spacer) region. The sequence of ITS region of the fungus MLJ-4 has been submitted to GenBank (Accession No. MZ930375.1). The strain has been deposited at the School of Pharmaceutical Sciences, Sun Yat-sen University.
4.3 Fermentation, extraction, and isolation
The fungus T. harzianum MLJ-4 was maintained on potato dextrose agar (PDA) medium at 28 ℃ for 5 d, and then three pieces (0.5 × 0.5 cm2) of mycelial agar plugs were inoculated into Erlenmeyer flasks, each containing 250 mL potato dextrose broth (PDB). After 4 days of incubation at 28 ℃ on a rotary shaker at 120 r/m, seed cultures were aseptically transferred into the rice solid medium (each flask contained 100.0 g of rice, 100 mL of natural filtered water). The 200 flasks were then kept at 28 ℃ for 30 days.
All fermentation products of the fungus MLJ-4 were exhaustively extracted with EtOAc (EA) for three times (each 20 L) at room temperature to afford 338.0 g of crude extract. Subsequently, the crude extract was subjected to silica gel (100−200 mesh) CC and eluted with mixed solvent of petroleum ether (PE) and EA to obtain 11 fractions (Frs. A−K).
Fr. E (4.6 g) was separated to CC on Rp-C18 silica gel and eluted with MeOH/H2O (40% → 100%) to led to seven sub-fractions (Frs. E1−E7). Fr. E5 (200.0 mg) was further purified by silica gel (300−400 mesh) CC (CH2Cl2/MeOH, 100:1 → 30:1) to obtain two sub-fractions (Frs. E5A and E5B). Fr. E5A (62.0 mg) was further purified by HPLC (MeCN/H2O, 50:50, 3 mL/min) to yield compounds 2 (3.2 mg, tR 18.5 min) and 7 (3.5 mg, tR 20.5 min). Fr. F (7.1 g) was subjected to CC on Rp-C18 silica gel and eluted with MeOH/H2O (40% → 100%) to give nine sub-fractions (Frs. F1−F9). Fr. F4 (419.0 mg) was further chromatographed by silica gel (300−400 mesh) CC (CH2Cl2/MeOH, 100:1 → 20:1) to obtain four sub-fractions (Fr. F4A−F4D). Fr. F4C (23.0 mg) was further purified by HPLC (MeCN/H2O, 65:35, 3 mL/min) to obtain compound 10 (5.6 mg, tR 11.0 min). Fr. G (29.6 g) was separated to CC on Rp-C18 silica gel and eluted with MeOH/H2O (30% → 100%) to give 11 sub-fractions (Frs. G1−G11). Fr. G11 (973.0 mg) was further separated by silica gel (300−400 mesh) CC (CH2Cl2/MeOH, 100:1 → 20:1) to obtain two sub-fractions (Frs. G11A and G11B). Fr. G11A (307.0 mg) was further purified by silica gel (300−400 mesh) CC (PE/EtOAc, 5:1 → 1:2) to obtain five sub-fractions (Frs. G11A1−G11A5). Fr. G11A1 (70.0 mg) was further purified by silica gel (300−400 mesh) CC (CH2Cl2/MeOH, 200:1 → 20:1) to obtain three sub-fractions (Frs. G11A1A−G11A1C), and compound 5 (3.6 mg, tR 14.0 min) was obtained from Fr. G11A1A (30.0 mg) by HPLC (MeCN/H2O, 50:50, 3 mL/min). Fr. G11A2 (40.0 mg) was further purified by HPLC (MeCN/H2O, 55:45, 3 mL/min) to afford compounds 3 (2.6 mg, tR 14.0 min), 6 (2.4 mg, tR 16.5 min), and 4 (2.9 mg, tR 17.5 min). Fr. H (41.0 g) was chromatographed over silica gel (300−400 mesh) (CH2Cl2/MeOH, 100:1 → 20:1) to obtain two sub-fractions (Frs. H1 and H2). Fr. H1 (21.4 g) was subjected to CC on Rp-C18 silica gel and eluted with MeOH/H2O (30% → 100%) to give 11 sub-fractions (Frs. H1A−H1K). Fr. H1G (690.0 mg) was further purified by silica gel (300−400 mesh) CC (CH2Cl2/MeOH, 100:1 → 20:1) to obtain four sub-fractions (Frs. H1G1A−H1G4D). Fr. H1G2B (20.0 mg) was further purified by HPLC (MeCN/H2O, 52:48, 3 mL/min) to obtain compound 1 (4.5 mg, tR 12.5 min). Fr. I (9.0 g) was subjected to CC on Rp-C18 silica gel and eluted with MeOH/H2O (30% → 100%) to give nine sub-fractions (Frs. I1−I9). Fr. I6 (1.3 g) was further purified by silica gel (300−400 mesh) CC (CH2Cl2/MeOH, 100:1 → 20:1) to obtain eight sub-fractions (Frs. I6A−I6H). Fr. I6F (179.0 mg) was further purified by HPLC (MeCN/H2O, 50:50, 3 mL/min) to give compounds 8 (4.9 mg, tR 12.5 min) and 9 (3.2 mg, tR 17.5 min).
4.4 Spectroscopic data of compounds
4.4.1 Harziachalasin A (1)
White amorphous powder; [α]D20 + 20 (c 0.1, MeOH); UV (MeCN) λmax (log ε) 190 (3.89) nm; ECD (c 0.22 × 10−3 M, MeCN) λmax (Δε) 190 (+ 12.87) nm; IR (UATR) νmax 3339, 2926, 1677, 1455, 1276, 1039, 750 cm−1; 1H and 13C NMR data see Table 1; HR-ESI–MS m/z 476.2384 [M + Na]+ (calcd. for C27H35NO5Na+, 476.2407).
4.4.2 Harziachalasin B (2)
White powder; [α]D20 + 38 (c 0.1, MeOH); UV (MeCN) λmax (log ε) 190 (4.01) nm; ECD (c 0.21 × 10−3 M, MeCN) λmax (Δε) 190 (+ 19.93), 228 (+ 3.35) nm; IR (UATR) νmax 2925, 1680, 1275, 1100, 906, 749 cm−1; 1H and 13C NMR data see Table 1; HR-ESI–MS m/z 500.2776 [M + Na]+ (calcd. for C30H39NO4Na+, 500.2771).
4.4.3 Harziachalasin C (3)
White powder; [α]D20 + 24 (c 0.1, MeOH); UV (MeCN) λmax (log ε) 190 (3.82) nm; ECD (c 0.20 × 10−3 M, MeCN) λmax (Δε) 190 (+ 21.87), 228 (+ 4.10) nm; IR (UATR) νmax 3355, 2924, 2854, 1679, 1455, 1373,1261, 1030, 908, 750 cm−1; 1H and 13C NMR data see Table 2; HR-ESI–MS m/z 516.2712 [M + Na]+ (calcd. for C30H39NO5Na+, 516.2720).
4.4.4 Harziachalasin D (4)
White powder; [α]D20 + 17 (c 0.1, MeOH); UV (MeCN) λmax (log ε) 190 (3.88) nm; ECD (c 0.19 × 10−3 M, MeCN) λmax (Δε) 190 (+ 16.23), 233 (+ 0.85) nm; IR (UATR) νmax 3348, 2926, 1679, 1556, 1496, 1454, 1376, 1262, 1027, 911, 737 cm−1; 1H and 13C NMR data see Table 2; HR-ESI–MS m/z 524.2988 [M + H]+ (calcd. for C31H42NO6+, 524.3007).
4.4.5 Harziachalasin E (5)
White powder; [α]D20 + 67 (c 0.1, MeOH); UV (MeCN) λmax (log ε) 190 (3.83) nm; ECD (c 0.21 × 10−3 M, MeCN) λmax (Δε) 190 (+ 14.43), 233 (+ 0.76) nm; IR (UATR) νmax 3337, 1639, 1275, 749 cm−1; 1H and 13C NMR data see Table 3; HR-ESI–MS m/z 488.2764 [M + Na]+ (calcd. for C29H39NO4Na+, 488.2771).
4.4.6 Harziachalasin F (6)
White powder; [α]D20−12 (c 0.1, MeOH); UV (MeCN) λmax (log ε) 190 (3.76) nm; ECD (c 0.23 × 10−3 M, MeCN) λmax (Δε) 190 (+ 12.35) nm; IR (UATR) νmax 3347, 2925, 2854, 1663, 1458, 1275, 1023, 748 cm−1; 1H and 13C NMR data see Table 3; HR-ESI–MS m/z 434.2681 [M + H]+ (calcd. for C28H36NO3+, 434.2690).
4.4.7 Harziachalasin G (7)
White powder; [α]D20 + 4 (c 0.1, MeOH); UV (MeCN) λmax (log ε) 190 (3.80) nm; ECD (c 0.21 × 10−3 M, MeCN) λmax (Δε) 192 (− 9.46), 227 (+ 2.14) nm; IR (UATR) νmax 3340, 2925, 1736, 1686, 1455, 1372, 1232, 1023, 750 cm−1; 1H and 13C NMR data see Table 3; HR-ESI–MS m/z 478.2943 [M + H]+ (calcd. for C30H40NO4+, 478.2952).
4.5 ECD calculations
The details of the quantum chemical ECD calculation for compounds 1−3 and 7 are provided in Supplementary Information (see S2).
4.6 Cell culture
The J-Lat A72 and J-Lat 10.6 cell lines (obtained from Professor Sun’ group at the School of Public Health, Shenzhen, Sun Yat-sen University), which serve as models for HIV latent infection, were maintained in RPMI 1640 medium (Gibco) containing 10% fetal bovine serum (Newzerum) and 1% Penicillin–Streptomycin (Gibco). HEK-293T cells were grown in DMEM medium supplemented with 10% fetal bovine serum and 1% Penicillin–Streptomycin. Cells were cultured at 37 ℃ in a humidified atmosphere with 5% CO2. Green fluorescent protein (GFP) expression was monitored as an indicator of HIV latency reversal following treatment.
4.7 HIV latency reversing assay
J-Lat A72 and J-Lat 10.6 cells were seeded at a density of 4 × 105 cells per well in 12-well plates and treated with test compounds for 24 h. Experimental controls included DMSO (negative control, NC) and 50 nM wikstroelide E (positive control, PC). Following treatment, cells were harvested by centrifugation (800 rpm, 5 min), washed with PBS, and analyzed for GFP expression. GFP-positive cells were visualized using an integrated cell imaging system (Thermo Fisher) and quantified by flow cytometry (Beckman).
4.8 Cell viability assay
The viability of HEK-293T cells was assessed using a Cell Counting Kit-8 (CCK-8) assay. Briefly, cells were plated in 96-well plates at a density of 10,000 cells per well in six replicates. Following a 24 h incubation, the cells were treated with a range of compound concentrations for another 24 h. After compound removal, CCK-8 reagent (GlpBio) was added according to the manufacturer's protocol, and the absorbance at 450 nm was measured after 3 h of incubation at 37 ℃.
4.9 RT-qPCR analysis
J-Lat A72 and J-Lat 10.6 cells were treated with DMSO (negative control, NC) or compound 4 for 24 h. Total RNA was extracted using TRIeasyTM Total RNA Extraction Reagent (Yeasen) and reverse transcribed into cDNA using Hifair® Ⅱ 1st Strand cDNA Synthesis SuperMix for qPCR (Yeasen). Quantitative RT-PCR was performed using Hieff® qPCR SYBR Green Master Mix (Yeasen) with the following primers sets:
GAPDH:
Forward: 5′-GTCTCCTCTGACTTCAACAGCG-3′.
Reverse: 5′-ACCACCCTGTTGCTGTAGCCAA-3′.
HIV gag:
Forward: 5′-AGCGTCAGTATTAAGCGGGG-3′.
Reverse: 5′-CAGGCCAGGATTAACTGCGA-3′.
HIV tat:
Forward: 5′-TAGACTAGAGCCCTGGAAGCA-3′.
Reverse: 5′-TCCGCTTCTTCCTGCCATAG-3′.
4.10 RNA-seq analysis
J-Lat 10.6 cells were treated with either DMSO (negative control, NC) or 10 μM compound 4 for 24 h. Total RNA was extracted and assessed for quality and concentration using a spectrophotometer or fluorometer. mRNA was purified from 1 μg of total RNA using oligo(dT) selection and used to synthesize double-stranded cDNA by SuperScript Double-Stranded cDNA Synthesis Kit (Invitrogen) with random hexamer primers. The cDNA underwent end-repair, phosphorylation, and adapter ligation. After purification and PCR amplification, the library was sequenced on the NovaSeq X Plus platform (PE150). Differential gene expression analysis was performed using DEGseq. Significantly differentially expressed genes were defined as those with |log2FC|≥ 1 and p-value < 0.05. Raw and processed RNA-seq data were deposited in NCBI GEO database with an accession number GSE303365.
4.11 Western blotting analysis
Cells treated with compound 4 under indicated conditions were collected by centrifugation (800 rpm, 25 ℃, 5 min) and lysed using IP buffer (Beyotime) containing phosphatase inhibitor (Selleck). Following centrifugation (12,000 rpm, 4 ℃, 20 min), the supernatant was mixed with 5 × dual color protein loading buffer and boiled at 100 ℃ for 5 min. Protein samples were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were blocked with 5% nonfat milk and incubated with overnight at 4 ℃ with primary antibodies against α-Tubulin (Zenbio, 250009, dilution ratio 1:4000), IκBα (Cell Signaling Technology, 44D4, dilution ratio 1:1000), and phospho-IκBα (Cell Signaling Technology, 14D4, dilution ratio 1:1000). After overnight incubation, the membranes were treated with appropriate secondary antibodies for visualization.
Notes
Acknowledgements
The authors thank the Science and Technology Program of Guangzhou, China (2024B03J1322), the Science and Technology Planning Project of Guangdong Province, China (2023A1111120025), the National Natural Science Foundation of China (82304322, 81973203, and 82404454), Jiangxi “Double Thousand Plan” Program (JXSQ2023102240), and the Guangdong Basic and Applied Basic Research Foundation, China (2021B1515140062) for providing financial support for this work.
Author contributions
YJZ and XYY designed and carried out the experiments and wrote the original manuscript; YFF and Q L assisted with data analysis and interpretation. JQC, XAC and DH conducted compound extraction, isolation, and structural characterization, and assisted in manuscript drafting; TY, XC, and SY assisted in analyzing the results; GHT designed the research framework and critically reviewed the entire manuscript. All authors read and approved the final manuscript.
Funding
The Science and Technology Program of Guangzhou, China (2024B03J1322), the Science and Technology Planning Project of Guangdong Province, China (2023A1111120025), National Natural Science Foundation of China (82304322, 81973203, and 82404454), Jiangxi “Double Thousand Plan” Program (JXSQ2023102240), the Guangdong Basic and Applied Basic Research Foundation, China (2021B1515140062).
Data availability
All data generated and analyzed during this study are included in this published article and its Additional file 1.
Declarations
Competing interests
The authors declare that there are no competing interests associated with this work.
References
-
1.Meng XF, Fang Y, Ding MY, Zhang YY, Jia KL, Li ZY, et al. Developing fungal heterologous expression platforms to explore and improve the production of natural products from fungal biodiversity. Biotechnol Adv. 2022;54: 107866. CrossRef PubMed Google Scholar
-
2.Tammam MA, Pereira F, Skellam E, Bidula S, Ganesan A, El-Demerdash A. The cytochalasans: potent fungal natural products with application from bench to bedside. Nat Prod Rep. 2025;42: 788-841. CrossRef PubMed Google Scholar
-
3.Scherlach K, Boettger D, Remme N, Hertweck C. The chemistry and biology of cytochalasans. Nat Prod Rep. 2010;27: 869-86. CrossRef PubMed Google Scholar
-
4.Skellam E. The biosynthesis of cytochalasans. Nat Prod Rep. 2017;34: 1252-63. CrossRef PubMed Google Scholar
-
5.Zhang JM, Liu X, Wei Q, Ma CT, Li DH, Zou Y. Berberine bridge enzyme-like oxidase-catalysed double bond isomerization acts as the pathway switch in cytochalasin synthesis. Nat Commun. 2022;13: 225. CrossRef PubMed Google Scholar
-
6.Chen R, Guo LJ, Li XD, Li XR, Hu K, Tang JW, et al. Phomopsischalins A–C, polycyclic-fused cytochalasins from the endophytic fungus Phomopsis sp. shj2 and their abilities to induce lysosomal function. Org Chem Front. 2023;10: 2218-25. CrossRef PubMed Google Scholar
-
7.Ding XM, Ye WX, Tan B, Song QY, Chen YC, Liu W, et al. Talachalasins A−C, undescribed cytochalasans with a 16β-methyl or 2-oxabicyclo[3.3.1]nonan-3-one unit from the deep-sea-derived fungus Talaromyces muroii sp. SCSIO 40439. Chin J Chem. 2023;41: 915-23. CrossRef PubMed Google Scholar
-
8.Hu XY, Li XM, Yang SQ, Li X, Wang BG, Meng LH. Vercytochalasins A and B: two unprecedented biosynthetically related cytochalasins from the deep-sea-sourced endozoic fungus Curvularia verruculosa. Chin Chem Lett. 2023;34: 107516. CrossRef PubMed Google Scholar
-
9.Yuan YY, Li Y, Lu WY, Liang AL, Li J, Wang WX. Xylariaides A and B, novel cytochalasans with a unique 5/6/5/3 ring system from a soil fungus Xylaria sp. Y01. Nat Prod Bioprospect. 2025;15: 23. CrossRef PubMed Google Scholar
-
10.Wang L, Yang J, Huang JP, Li J, Luo JY, Yan YJ, et al. Bisaspochalasins A-C: three cytochalasan homodimers with highly fused ring system from an endophytic Aspergillus flavipes. Org Lett. 2020;22: 7930-5. CrossRef PubMed Google Scholar
-
11.Wang L, Yu ZY, Guo XW, Huang JP, Yan YJ, Huang SX, et al. Bisaspochalasins D and E: two heterocycle-fused cytochalasan homodimers from an endophytic Aspergillus flavipes. J Org Chem. 2021;86: 11198-205. CrossRef PubMed Google Scholar
-
12.Wang WJ, Zeng FR, Bie Q, Dai C, Chen CM, Tong QY, et al. Cytochathiazines A-C: three merocytochalasans with a 2H–1,4-thiazine functionality from coculture of Chaetomium globosum and Aspergillus flavipes. Org Lett. 2018;20: 6817-21. CrossRef PubMed Google Scholar
-
13.Ma KL, Dong SH, Li HY, Wei WJ, Tu YQ, Gao K. Cytochalasins from Xylaria sp. CFL5, an endophytic fungus of Cephalotaxus fortunei. Nat Prod Bioprospect. 2021;11: 87-98. CrossRef PubMed Google Scholar
-
14.Wu ZD, Zhang XT, Chen CM, Zhou P, Zhang M, Gu LH, et al. Dimericchalasine A and amichalasines D and E: unexpected cytochalasan homodimer and heterotrimers from Aspergillus micronesiensis PG-1. Org Lett. 2020;22: 2162-6. CrossRef PubMed Google Scholar
-
15.Wu ZD, Zhang XT, Li Q, Tong QY, Yang J, Chen CM, et al. Amichalasines F-J: cytochalasan heterotrimers with mirror-imaged core structures from Aspergillus micronesiensis. Org Chem Front. 2023;10: 3530-6. CrossRef PubMed Google Scholar
-
16.Zhou JT, Wu Q, Zhao JX, Wu LL, He XH, Liang LQ, et al. Sucurchalasins A and B, sulfur-containing heterodimers of a cytochalasan and a macrolide from the endophytic fungus Aspergillus spelaeus GDGJ-286. J Nat Prod. 2024;87: 2327-34. CrossRef PubMed Google Scholar
-
17.Zhang XT, Yang L, Wang WJ, Wu ZD, Wang JP, Sun WG, et al. Flavipesines A and B and asperchalasines E-H: cytochalasans and merocytochalasans from Aspergillus flavipes. J Nat Prod. 2019;82: 2994-3001. CrossRef PubMed Google Scholar
-
18.Kim M, Song K, Jin EJ, Sonn J. Staurosporine and cytochalasin D induce chondrogenesis by regulation of actin dynamics in different way. Exp Mol Med. 2012;44: 521-8. CrossRef PubMed Google Scholar
-
19.Jiang TT, Lee JW, Collins JE, Schaefer S, Chen D, Nardella F, et al. Fungal-derived methyldeoxaphomins target Plasmodium falciparum segregation through the inhibition of PfActin1. Proc Natl Acad Sci U S A. 2025;122: e2418871122. CrossRef PubMed Google Scholar
-
20.Xu CR, Shi YH, Zhang WJ, Yang JF, Zhang XF, Chen SB, et al. Derailment of the biosynthesis via an acid-mediated intramolecular cyclo-rearrangement leads to a novel cytochalasin skeleton. J Nat Prod. 2025;88: 477-84. CrossRef PubMed Google Scholar
-
21.Long XW, Ding YM, Deng J. Total synthesis of asperchalasines A, D, E, and H. Angew Chem Int Ed. 2018;57: 14221-4. CrossRef PubMed Google Scholar
-
22.Wu H, Ding YM, Hu K, Long XM, Qu CL, Puno PT, et al. Back cover: bioinspired network analysis enabled divergent syntheses and structure revision of pentacyclic cytochalasans. Angew Chem Int Ed. 2021;60: 16240. CrossRef PubMed Google Scholar
-
23.Osborne O, Peyravian N, Nair M, Daunert S, Toborek M. The paradox of HIV blood–brain barrier penetrance and antiretroviral drug delivery deficiencies. Trends Neurosci. 2020;43: 695-708. CrossRef PubMed Google Scholar
-
24.Loucif H, Gouard S, Dagenais-Lussier X, Murira A, Stäger S, Tremblay C, et al. Deciphering natural control of HIV-1: a valuable strategy to achieve antiretroviral therapy termination. Cytokine Growth Factor Rev. 2018;40: 90-8. CrossRef PubMed Google Scholar
-
25.Marsden MD, Zhang TH, Du YS, Dimapasoc M, Soliman MSA, Wu XM, et al. Tracking HIV rebound following latency reversal using barcoded HIV. Cell Rep Med. 2020;1: 100162. CrossRef PubMed Google Scholar
-
26.Sloane JL, Benner NL, Keenan KN, Zang XY, Soliman MSA, Wu XM, et al. Prodrugs of PKC modulators show enhanced HIV latency reversal and an expanded therapeutic window. Proc Natl Acad Sci U S A. 2020;117: 10688-98. CrossRef PubMed Google Scholar
-
27.Sadowski I, Hashemi FB. Strategies to eradicate HIV from infected patients: elimination of latent provirus reservoirs. Cell Mol Life Sci. 2019;76: 3583-600. CrossRef PubMed Google Scholar
-
28.Chen CM, Zhu HC, Wang JP, Yang J, Li XN, Wang J, et al. Armochaetoglobins K-R, anti-HIV pyrrole-based cytochalasans from Chaetomium globosum TW1-1. Eur J Org Chem. 2015;2015: 3086-94. CrossRef PubMed Google Scholar
-
29.Zhang YG, Tian RR, Liu SC, Chen XL, Liu XZ, Che YS. Alachalasins A–G, new cytochalasins from the fungus Stachybotrys charatum. Bioorg Med Chem. 2008;16: 2627-34. CrossRef PubMed Google Scholar
-
30.Chen JQ, Li S, Fan RZ, Sun ZH, Zhu XY, Yin AP, et al. Talaesthanes A-C, three new meroterpenoids from the endophytic fungus Talaromyces primulinus H21. Fitoterapia. 2024;177: 106085. CrossRef PubMed Google Scholar
-
31.Li L, Zhu XY, Zhu JY, Cui QY, Lin YW, Lu QR, et al. HSQC-guided discovery and structural optimization of antiadipogenic indole diterpenoids from endophytic fungus Penicillium janthinellum H-6. Eur J Med Chem. 2025;297: 117956. CrossRef PubMed Google Scholar
-
32.Li L, Zhu XY, Zhu JY, Wu ZH, Zhang YJ, Yuan FY, et al. Penispirolactam and penipyrroloindole, two unusual polycyclic indole diterpenoids with anti-hepatic fibrosis activity from the endophytic fungus Penicillium janthinellum H-6. Chin Chem Lett. 2025. CrossRef PubMed Google Scholar
-
33.Lu QR, Li L, Cui QY, Liao Q, Malik N, Wu LM, et al. Sclerotiorin-type azaphilones isolated from a marine-derived fungus Microsphaeropsis arundinis P1B. J Nat Prod. 2025;88: 1075-84. CrossRef PubMed Google Scholar
-
34.Wang Y, Song SH, Wu LM, Zhou X, Lu QR, Yin AP, et al. Chemical constituents of Penicillium ferraniaense GE-7 and their cytotoxicities. Nat Prod Res. 2025;39: 3915-22. CrossRef PubMed Google Scholar
-
35.Weng HZ, Zhu JY, Yuan FY, Tang ZY, Tian XQ, Chen Y, et al. Homo/Hetero-dimers of aromatic bisabolane sesquiterpenoids with neuroprotective activity from the fungus Aspergillus versicolor A18 from south China sea. Mar Drugs. 2022;20: 322. CrossRef PubMed Google Scholar
-
36.Wu SQ, Zhu XY, Yuan FY, Weng HZ, Li L, Huang D, et al. Malbrumpenoids A-N, unusually cyclized triterpenoids from the Euphorbia endophyte Malbranchea umbrina D16. Chin Chem Lett. 2025. CrossRef PubMed Google Scholar
-
37.Wu WY, Wei X, Liao Q, Fu YF, Wu LM, Li L, et al. Structurally diverse polyketides and alkaloids produced by a plant-derived fungus Penicillium canescens L1. Nat Prod Bioprospect. 2025;15: 22. CrossRef PubMed Google Scholar
-
38.Gao SY, Wu P, Xue JH, Li HX, Wei XY. Cytochalasans from the endophytic fungus Diaporthe ueckerae associated with the fern Pteris vittata. Phytochemistry. 2022;202: 113295. CrossRef PubMed Google Scholar
-
39.Jordan A, Bisgrove D, Verdin E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003;22: 1868-77. CrossRef PubMed Google Scholar
-
40.Li SF, Liang X, Wu XK, Gao X, Zhang LW. Discovering the mechanisms of wikstroelide E as a potential HIV-latency-reversing agent by transcriptome profiling. J Nat Prod. 2021;84: 1022-33. CrossRef PubMed Google Scholar
-
41.Griffin GE, Leung K, Folks TM, Kunkel S, Nabel GJ. Activation of HIV gene expression during monocyte differentiation by induction of NF-κB. Nature. 1989;339: 70-3. CrossRef PubMed Google Scholar
-
42.Nixon CC, Mavigner M, Sampey GC, Brooks AD, Spagnuolo RA, Irlbeck DM, et al. Systemic HIV and SIV latency reversal via non-canonical NF-κB signalling in vivo. Nature. 2020;578: 160-5. CrossRef PubMed Google Scholar
-
43.Wu ZY, Ding K, Liu WL, He H, Chen MY, Chen MX, et al. Wikstrol B reactivates latent human immunodeficiency virus (HIV-1) via the nuclear factor-κB (NF-κB) pathway. Phytomedicine. 2025;141: 156667. CrossRef PubMed Google Scholar
-
44.Chang HT, Chou CT, Chen IS, Yu CC, Lu T, Hsu SS, et al. Mechanisms underlying effect of the mycotoxin cytochalasin B on induction of cytotoxicity, modulation of cell cycle, Ca2+ homeostasis and ROS production in human breast cells. Toxicology. 2016;370: 1-19. CrossRef PubMed Google Scholar
-
45.Peng XG, He YZ, Gao Y, Duan FF, Chen J, Ruan HL. Cytochalasins from an endophytic fungus Phoma multirostrata XJ-2-1 with cell cycle arrest and TRAIL-resistance-overcoming activities. Bioorg Chem. 2020;104: 104317. CrossRef PubMed Google Scholar
-
46.Zhang GH, Liu J, Li SZ, Wang TY, Chen L, Li H, et al. Cytochalasin H enhances sensitivity to gefitinib in non-small-cell lung cancer cells through inhibiting EGFR activation and PD-L1 expression. Sci Rep. 2024;14: 25276. CrossRef PubMed Google Scholar
-
47.Lurain K. Treating cancer in people with HIV. J Clin Oncol. 2023;41: 3682-8. CrossRef PubMed Google Scholar
-
48.Engels EA, Yanik EL, Wheeler W, Gill MJ, Shiels MS, Dubrow R, et al. North American AIDS cohort collaboration on research and design of the international epidemiologic databases to evaluate AIDS; Cancer-attributable mortality among people with treated human immunodeficiency virus infection in north America. Clin Infect Dis. 2017;65: 636-43. CrossRef PubMed Google Scholar
-
49.Goehringer F, Bonnet F, Salmon D, Cacoub P, Paye A, Chêne G, et al. Causes of death in HIV-infected individuals with ommunovirologic success in a national prospective survey. AIDS Res Hum Retroviruses. 2017;33: 187-93. CrossRef PubMed Google Scholar
-
50.Yang L, Wang Q, Ma QY, Xie QY, Gai CJ, Wu YG, et al. Diaporchalasins A–E, new cytochalasins from the endophytic fungus Diaporthe sp. BMX12 isolated from Aquilaria sinensis. Chem Biodivers. 2024;21: e202400567. CrossRef PubMed Google Scholar
-
51.Wang WX, Cheng GG, Li ZH, Ai HL, He J, Li J, et al. Curtachalasins, immunosuppressive agents from the endophytic fungus Xylaria cf. curta. Org Biomol Chem. 2019;17: 7985-94. CrossRef PubMed Google Scholar
Copyright information
© The Author(s) 2026.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
