
Panunoids A – D, four new prenylhydroquinone derivatives isolated from the fungus Panus rudis
Abstract
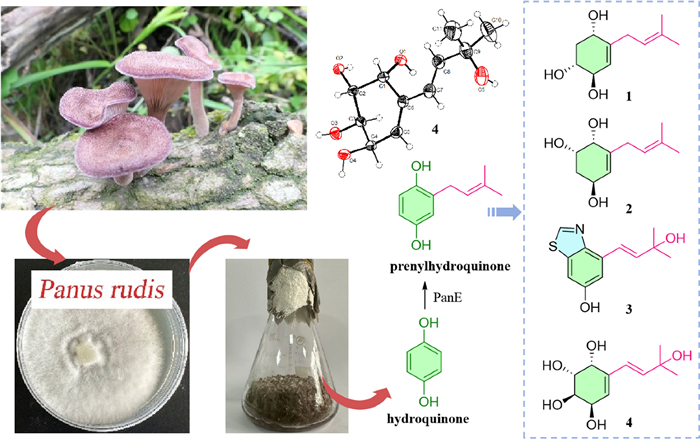
A chemical constituent study on the fermented rice substrate of basidiomycetous fungus Panus rudis led to the isolation of four previously undescribed prenylhydroquinone derivatives compounds (1–4) and eight known compounds (5–12). Among them, compound 3 featured a rare benzothiazole derivative with hydroxy substituted 3-methyl-1-butenyl substitution on the benzene ring, and the absolute configurations of 7 and 12 were elucidated as unreported ones. Their structures were identified by the interpretation of 1D and 2D NMR spectroscopy, HRESIMS data, X-ray single-crystal diffraction, and comparison of calculated and experimental ECD spectra. The plausible biosynthetic pathways for 1–7 are proposed. Cytotoxicity evaluation was conducted on 1 and 3–12 against two cancer cell lines (A-549 and HepG2). The results demonstrated that 1, 3–6, 8, 9, and 12 exhibited weak cytotoxicity against both two cell lines. Among them, 8 and 12 showed dose-dependent inhibitory effects, and their IC50 values at 72 h were obtained.Graphical Abstract
Keywords
Panus rudis Solid-state fermentation Benzothiazole derivatives Cytotoxicity1 Introduction
Panus rudis (Polyporales, Panaceae) shows remarkable economic and ecological value by secretion of lignin-modifying enzymes (e.g., laccase) [1–3]. And it is a lignicolous fungus belonging to Basidiomycota, which comprises most mushroom-type fungi and is recognized as an ingenious producer of highly functionalized natural products and a rich source of biosynthetic enzymes [4]. Panepoxydone, biosynthesized from prenylhydroquinone through successive hydroxylation, epoxidation, and reduction reactions, was first isolated from P. rudis, which exhibited multiple biological activities, such as inhibitory activity against NF-κB as well as antitumor activity [5]. Afterwards, a series of panepoxydone analogues were obtained through liquid fermentation or precursor-directed biosynthesis by P. rudis [6–8]. Hexacyclinol, a dimeric prenylhydroquinone isolated from this genus, had been highly controversial. Its accurate structure was ultimately established through the calculation of the13C-NMR chemical shifts. Subsequently, Porco et al. completed the total synthesis of the reassigned (+)-hexacyclinol and confirmed its absolute configuration via X-ray crystallography [9–11]. In recent years, structurally diverse dimers and their respective monomeric intermediates had even been proven to be produced by P. rudis, using 5-FAM-maleimide combined with UHPLC–MS/MS–FBMN workflow. And dimers with four types of complexes and densely functionalized cores were identified from this fungus [12]. Moreover, abundant secondary metabolites also have been reported from the fungi of Panus [13–15]. Recently, four racemic cyclohexene dimers and two non-racemic cyclohexene monomers were isolated from P. similis [16]. Among them, (3R*,4S*)-2,2-Dimethyl-3,4,6-trihydroxychromane exhibited selective cytotoxicity towards NCI-H187 cells (IC50 = 9.1 μM). However, most Panus fungi had been investigated for their fermentation broths, but the metabolic potential of P. rudis within solid-state fermentation systems remains to be further explored. Solid-state fermentation, as a time-honored bioprocessing technology, plays an indispensable role in the production of characteristic fermented foods which not only confers distinctive flavors and textures to food products but also biosynthesizes functional secondary metabolites [17–20]. And rice as a solid-state fermentation medium shows the great potential of discovering more natural metabolites with diverse structures or significant bioactivities [21–23].
This study investigating the rice-based solid-state fermentation metabolites of P. rudis by employing multifaceted chromatographic separation coupled with spectroscopic characterization techniques resulted in the isolation of a series of prenylhydroquinone derivatives (Fig. 1), and the plausible biosynthetic pathway of them was inferred. In order to screen compounds with potential for biological activities, the cytotoxicity of the isolates was also tested.
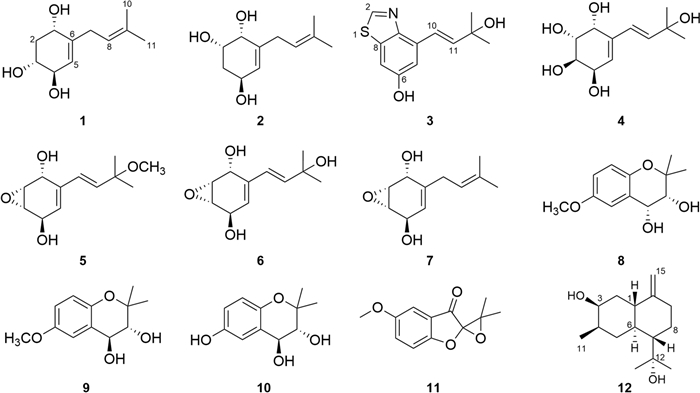
The structures of compounds 1–12
2 Results and discussion
Compound 1 was obtained as a white amorphous powder, and its molecular formula was determined as C11H18O3 by means of HR-ESI–MS (m/z 221.1151 [M + Na]+, calcd for C11H18O3Na+, m/z 221.1154). The 1H NMR spectrum data (Table 1) indicated the presence of two olefinic methines at δH 5.30 (m, 1H) and 5.19 (m, 1H), three oxygenated methines at δH 4.19 (m, 1H), 3.98 (m, 1H) and 3.49 (ddd, J = 11.8, 7.5, 3.4 Hz, 1H), two methylene at δH 2.81 (m, 2H), 2.23 (ddd, J = 12.3, 5.7, 3.5 Hz, 1H) and 1.60 (overlapped, 1H), and two methyl groups at δH 1.73 (s, 3H) and 1.63 (s, 3H). 13C NMR (Table 1) and HSQC spectrum revealed the signals of 11 carbons, including two double bonds (δC 143.5, 134.4, 126.0 and 122.6), three oxygenated methines (δC 74.3, 73.2 and 69.3), two methylenes (δC 40.5 and 31.7), and two methyls (δC 17.9 and 26.1). Two structural fragments (C-1/C-2/C-3/C-4/C-5 and C-7/C-8) can be readily deduced on the basis of 1H–1H COSY spectrum (Fig. 2). The HMBC correlations (Fig. 2) between the two methyl groups and the olefinic methine indicate the presence of an isopentenyl moiety. According to the HMBC cross-peaks from H-5 to C-3/C-1, from H-2 to C-4/C-6, and from H-1 to C-5, combined with 1H–1H COSY correlations (H-1/H-2/H-3/H-4/H-5), a cyclohexene fragment with hydroxyl groups can be constructed. The connectivity of the above two fragments through C-6 and C-7 was established based on the HMBC correlations from H-7 to C-1/C-5 and from H-5 to C-7. Based on the aforementioned NMR data, the planar structure of 1 was characterized (Fig. 1).
1H and 13C NMR Data of Compounds 1–4
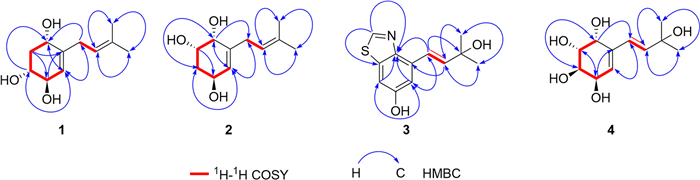
Key 1H-1H COSY and HMBC correlations of compounds 1–4
The NOESY correlations (Fig. 3) of H-1 with H-3, which indicated that H-1 and H-3 exhibit a cis-configuration. By further interpreting the multiplicities of H-3 (J3,2a = 11.8, J3,2e = 3.4, J3,4 = 7.5), the anti-orientation of H-3/H-4 was suggested by a large J3,4 (7.5 Hz). Thus, the relative configuration of 1 can be determined as shown in Fig. 3. Its absolute configuration was speculated to be 1S,3R,4R upon comparing its experimental ECD curve (Fig. 4) with the calculated ones, and 1 was named as panunoid A.
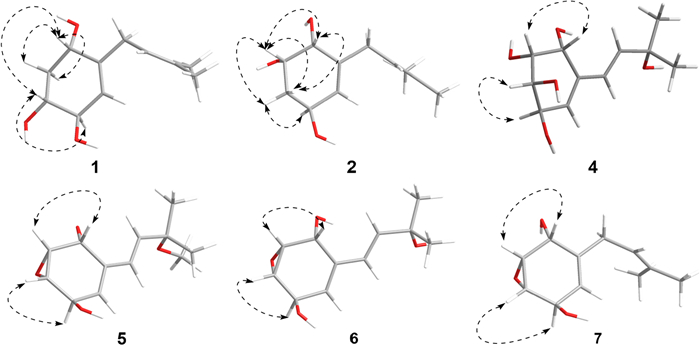
Key NOESY correlations of compounds 1, 2, and 5–7 and key ROESY correlations of compound 4
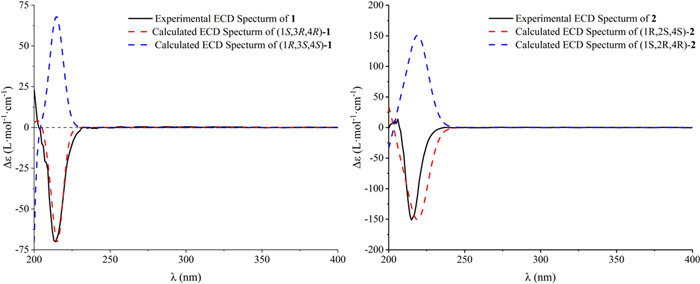
Experimental and calculated ECD spectra of compounds 1 and 2
Compound 2 was obtained as a white amorphous powder, and its molecular formula was determined as C11H18O3 the same with 1, by means of HR-ESI–MS (m/z 199.1326 [M + H]+, calcd for C11H19O3+, m/z 199.1334). The 1H and 13C NMR data of 2 (Table 1) closely resembled those of 1. The differences in carbon chemical shifts were observed at the hydroxyl-substituted cyclohexene moieties, indicating that they could be isomers differing in the positions or configurations of the hydroxyl groups. The planar structure of 2 was further confirmed through 2D NMR spectra (Fig. 2).
The NOESY correlations (H-1/H-2) and the small J1,2 (3.5) of compound 2 suggested the cis-orientation of H-1/H-2. By comparing with 1, no correlation between H-2 and H-4 was observed, it can be presumed that H-2 and H-4 are arranged in a trans configuration. Thus, the relative configuration of 2 could be speculated as shown in Fig. 3. The possible absolute configuration was supposed to be 1R,2S,4S upon comparing its experimental ECD curve (Fig. 4) with the calculated ones and 2 (Fig. 1) was named as panunoid B.
Compound 3 was obtained as a brown oil, and its molecular formula was determined as C12H13NO2S by means of HR-ESI–MS (m/z 234.0596 [M–H]–, calcd for C12H12NO2S–, m/z 234.0589; m/z 218.0644 [M–H2O + H]+, calcd for C12H12NOS+, m/z 218.0640). The 1H NMR spectrum data (Table 1) indicated the presence of two coupled protons on the double bond at δH 7.30 (d, J = 16.2 Hz, 1H) and 6.70 (d, J = 16.2 Hz, 1H), along with two coupled aromatic protons [δH 7.25 (d, J = 2.3 Hz, 1H) and 7.15 (d, J = 2.3 Hz, 1H)] at the meta-position on the benzene ring. Additionally, a deshielded aromatic proton signal was observed at δH 8.93 (s, 1H), which may be attributed to an aromatic heterocyclic group. Moreover, two methyl groups in the same chemical environment at δH 1.44 (s, 6H) were observed in 1H NMR spectrum. The 13C NMR (Table 1) and HSQC spectrum data revealed the signals of 12 carbons, including an oxygenated quaternary carbon at δC 71.8, a double bond at δC 141.9 and 123.7, two methyl carbons resonating at δC 30.0, along with other seven aromatic carbons distributed between δC 157.6 and 106.9.
Based on the HMBC correlations from H-2 to C-8/C-9, H-5 to C-7/C-9, and from H-7 to C-9/C-5 combined with its molecular formula, the partial structure of 6-hydroxybenzothiazole could be obtained, which was also reinforced by comparing with the NMR data reported in the literature [24]. Furthermore, the correlations from H-10 to C-5/C-9/C-12, H-5 to C-10, H-11 to C-4, and H-13 to C-11 indicated that a hydroxy substituted 3-methyl-1-butenyl was connected with C-4. In summary, the structure of 3 can be obtained as drawn in Fig. 1 and named as panunoid C.
Compound 4 was obtained as a colorless crystal. The 1H NMR spectra were recorded both in CD3OD and DMSO-d6 (Table 1), and the presence of hydroxyl signals resulted in more complex coupling patterns for hydrogen signals recorded in DMSO-d6 than those recorded in CD3OD. The 1H NMR spectrum recorded in CD3OD showed the presence of an olefinic methine at 5.60 (d, J = 4.8 Hz, 1H), two trans-coupled hydrogens on the double at δH 6.20 (d, J = 16.1 Hz, 1H) and 6.12 (d, J = 16.1 Hz, 1H), four oxygenated methines at δH 4.42 (d, J = 4.0 Hz, 1H), 4.27 (d, J = 4.8 Hz, 1H), 3.85 (dd, J = 10.5, 4.4 Hz, 1H) and 3.77 (dd, J = 10.5, 4.0 Hz, 1H), and two methyls at δH 1.31 (s, 3H) and 1.30 (s, 3H). 13C NMR (Table 1) and HSQC spectra revealed the signals of 11 carbons, including two double bonds at δC 137.3, 139.5, 128.5 and 125.7, four oxygenated methine carbons at δC 69.0, 68.4, 66.6 and 66.3, an oxygenated quaternary carbon at δC 69.2, and two methyls at δC 30.2 and 30.1, respectively.
The 1H–1H COSY spectrum (Fig. 2) determined two partial structures of C-1/C-2/C-3/C-4/C-5 and C-7/C-8, which showed similarity to those of 1. Then the planar structure of 4 was established as shown in Fig. 1 through HMBC correlations from H-10/H-11 to C-8, H-8 to C-10/C-11, H-7 to C-5, H-5 to C-7, and H-1 to C-7/C-3. The NOESY cross-peaks of H-1/H-2 and H-3/H-4 (Fig. 3) together with the small J1,2 (4.0) and J3,4 (4.4), and the big J2,3 (10.5) revealed that H-1 and H-2 were on the same side of the ring, so were H-3 and H-4. Furthermore, the absolute configuration of 4 (Deposition No. CCDC-2417279) was determined by single-crystal X-ray diffraction (Cu Kα) with a Flack parameter 0.03(4) (Fig. 5) and named as panunoid D.
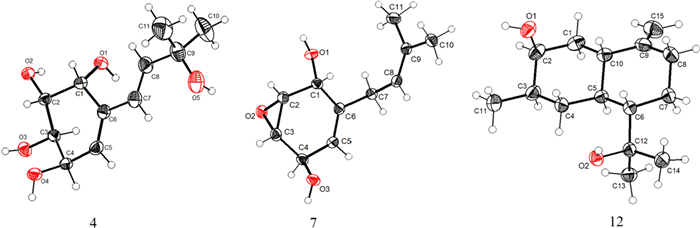
ORTEP drawing of 4, 7, and 12 obtained by X-ray analysis
Molecular formula of compound 7 was determined as C11H16O3 by means of HR-ESI–MS (m/z 197.1174 [M + H]+, calcd for C11H17O3+, m/z 197.1178). The 1H NMR and 13C NMR data see Table 2. The planar structure, as determined by 1D and 2D NMR spectroscopy, was consistent with that of 7-desoxyphenepoxydol [25]. The NOESY spectrum (Fig. 3) revealed correlations of H-1/H-2 and H-3/H-4, with the cross-peak intensity of H-1/H-2 being significantly stronger than that of H-3/H-4. Analogous characteristics of intensity of cross-peaks (H-1/H-2 and H-3/H-4) were also observed in the 1H–1H COSY spectrum. This observation suggested a cis-configuration between H-1 and H-2. Combining with the above-mentioned information and the absence of correlation between H-1 and H-4 suggested that the epoxide and hydroxyl group of C-1 were syn-oriented and the epoxide and hydroxyl group of C-4 were anti-oriented. By single-crystal X-ray diffraction (Cu Kα) with a Flack parameter 0.04(13) (Fig. 5), its absolute configuration was determined as 1R,2R,3S,4R.
1H and 13C NMR Data of Compounds 5–7
The molecular formula of 5 and 6 was determined as C12H18O4 and C11H16O4 by means of HR-ESI–MS (m/z 249.1119 [M + Na]+, calcd for C12H18NaO4+, m/z 249.1097 and m/z 235.1056 [M + Na]+, calcd for C11H16NaO4+, m/z 235.0941), respectively. The 1H NMR and 13C NMR data (Table 2) of 5 were consistent with panepoxydiol [6]. The NMR data (Table 2) of 6 were similar to those of 5, except that the methoxy group (C-9) was replaced by a hydroxyl group, thus the planar structure of 6 was the same with (E)-3-(3-hydroxy-3-methylbut-1-en-1-yl)-7-oxabicyclo[4.1.0]hept-3-ene-2,5-diol [6, 26]. Lentinoid B was previously reported with the same planar structure of 4 but the same spectra data with 6 [27]. The structural revision of the lentinoid B into compound 6 was determined based on its chemical shift of C-2 (δC 57.1) and C-3 (δC 56.0) being upfield-shifted compared to compound 4 (C-2 δC 68.4, C-3 δC 69.0) and resembling that of other epoxide analogues. The correlations and the features of cross-peak intensity in NOESY and COSY spectra for both 5 and 6 were consistent with those of 7. The peak shapes and coupling constants in the 1H NMR spectrum (Table 2) of 5, 6 and 7 are identical. Consistency between spectroscopic data and biosynthetic logic suggested that compounds 5, 6, and 7 shared an identical stereochemical configuration (Fig. 1).
The other known compounds were identified as (3R,4R)-3,4-dihydroxy-6-methoxy-2,2-dimethylchroman (8) [28], (3R,4S)-3,4-dihydroxy-6-methoxy-2,2-dimethylchroman (9) [29], (3R,4S)-3,4,6-dihydroxy-2,2-dimethyl-chroman (10) [30], 5-Methoxy-3′,3′-dimethyl-3H-spiro[benzofuran-2,2′-oxiran]-3-one (11) [15] by comparison of NMR spectroscopic data with published literature values. Among them, 11 might be a racemic mixture. (4S)-3α,12-Dihydroxycadin-10(15)-ene previously obtained by biotransformation of cadina-4,10(15)-dien-3-one, a substrate added exogenously in the culture medium, by the fungi Mucor plumbeus ATCC 4740 [31] shared the same planar structure with 12. In this work, its absolute configuration was determined as 1S,3S,4R,6S,7S by single-crystal X-ray diffraction (Cu Kα) with a Flack parameter 0.04(4) (Fig. 5).
The plausible biosynthetic pathway for compounds 1–7 (Fig. 6) was proposed starting from hydroquinone [5]. The premise of the whole process is to modify hydroquinone by PanE to obtain intermediate i. Briefly, compounds 1 and 2 were derived from intermediate i by oxidation, hydroxylation, and reduction, respectively [32]. The intermediate ii was catalytically epoxidated by PanH and subsequently reduced to obtain compound 7. What is more, intermediate i was modified by monooxygenase, isomerized and oxidized to produce intermediate iii. The condensation of cysteine with intermediate iii afforded intermediate v. Compound 3 was obtained from intermediate v through a series of steps [33, 34]. Intermediate iv was obtained by catalytic epoxidation of intermediate iii via PanH. Intermediate iv was reduced to give compound 6. In addition, compounds 5 and 4 were obtained from compound 6 through methoxylation and hydrolysis, respectively.
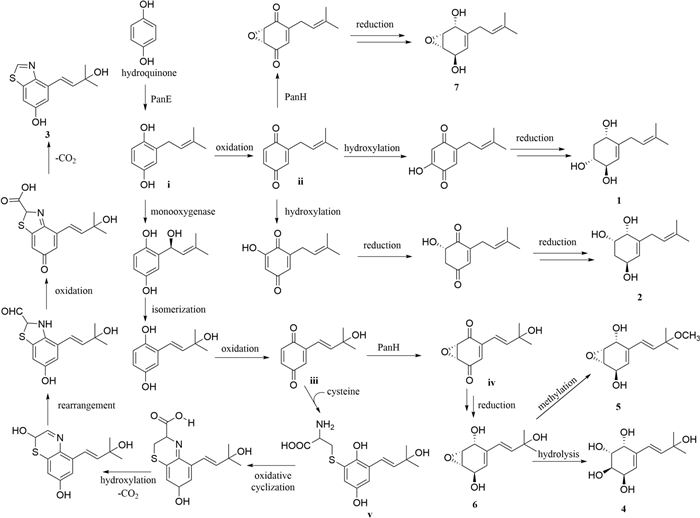
The plausible biosynthetic pathway for compounds 1–7
All compounds were subjected to cytotoxicity testing, except for 2, which had a small amount. The in vitro activity results revealed that 1, 3–6, 8, 9, and 12 exhibited cytotoxic effects against A-549 and HepG2 cell lines (Fig. 7). Among them, 8 and 12 have stronger cell viability inhibitory effects on A-549 and HepG2 cells, and show a dose-dependent effect (Fig. 7). Compound 8 has an IC50 value of 64.27 μg/mL against A-549 cells and 75.61 μg/mL against HepG2 cells. Compound 12 has an IC50 value of 79.09 μg/mL against A549 and 103.5 μg/mL against HepG2.
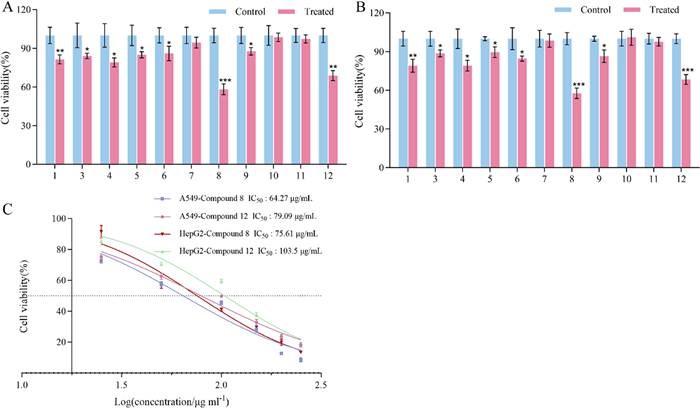
Effect on cell viability of A-549 and HepG2 in vitro. A. Cell viability of A-549 after 72 h of drug treatment (concentration: 50 μg/mL); B. Cell viability of HepG2 after 72 h of drug treatment (concentration: 50 μg/mL); and C. IC50 values after 72 h of compound treatment (concentrations: 250, 200, 150, 100, 50, and 25 μg/mL). Note: P < 0.05, **P < 0.01, ***P < 0.001
3 Experimental
3.1 General
A KRÜSS P8000 (KRÜSS GmbH, Hamburg, Germany) automatic polarimeter were used to measure specific rotations. A MOS-500 (Konica Minolta Sensing, Inc., Osaka, Japan) spectrometer was used to measure CD spectra. HR-ESI–MS was determined using Acquity UPLC I-class plus tandem Xevo G2-XS Qtof (Waters Co., Milford, MA, USA). Single crystal X-ray structure was obtained on Bruker D8 Venture (Bruker AXS GmbH, Karlsruhe, Germany). 1D and 2D NMR spectra were measured on Bruker Avance-400 and Avance-600 (Bruker BioSpin GmbH, Germany) spectrometers with CD3OD and DMSO-d6 as solvents and TMS as internal standard. Column chromatography (CC) was carried out over silica gel (200–300 mesh) (Qingdao Haiyang Chemical Co., Qingdao, China). Pre-coated silica gel plates (Qingdao Haiyang Chemical Co., Qingdao, China) were used for thin-layer chromatography (TLC). Detection was done under UV light (254 and 365 nm) and by spraying the plates with 10% sulfuric acid ethanol solution followed by heating. An Agilent series 1260 (Agilent Technologies, Santa Clara, CA, USA) was used for analysis HPLC. Semi-preparative HPLC was done on a Hanbon series NP7005C (Hanbon Sci & Tech, Jiangsu, China), the column used was HB-C18FP-100-9 (9 μm, 100Å, 20 × 250 mm, Hanbon Sci & Tech, Jiangsu, China). A CHIRALPAK® AD-H column (5 μm, 4.6 × 250 mm, Daicel Corporation, Japan) was employed for chiral compound separation.
3.2 Fungi culture
P. rudis strain CGMCC 5.34 was purchased from China General Microbiological Culture Collection Center, and stored in Jiangsu Key Laboratory of Regional Specific Resource Pharmaceutical Transformation. The strain was cultured on YMG solid medium and incubated at 28 ℃ for mycelia production. Subsequently, the mycelia were aseptically transferred into conical flasks containing YMG liquid medium. These were incubated as seed cultures for 7 days on a rotary shaker at 130 rpm (28 ℃). For large-scale cultivation, thirty 500 mL conical flasks were prepared, each containing 100 g rice supplemented with 100 mL purified water. Each flask was inoculated with 10 mL of seed medium and incubated statically at 28 ℃ for 40 days. The fermented rice substrate was subjected to methanol for extraction (3 × 20 L × 3 days). After combining the extracts, the organic solvent was removed via rotary evaporation under vacuum, yielding 2.4 L of concentrated aqueous solution. This solution was extracted three times with ethyl acetate, and the combined organic phases were concentrated to dryness under vacuum, ultimately obtaining 12.76 g of crude extract.
3.3 Extraction and isolation
The crude extract (12.76 g) was submitted to silica gel (200–300 mesh) column chromatography (CC), eluting with petroleum ether–EtOAc gradient system (100:0, 50:1, 30:1, 20:1, 10:1, 8:1, 5:1, 1:1, v/v), and then eluting with CH2Cl2–MeOH gradient system (20:1, 15:1, 10:1, 9:1, 8:2, 7:3, 6:4, 1:1, 0:100, v/v), to yield nine fractions (L1–L9).
L4 (3.3 g) was applied to silica gel (200–300 mesh) CC, and eluting with petroleum ether–EtOAc gradient system (100:0, 30:1, 20:1, 10:1, 8:1, 5:1, 1:1, v/v) and CH2Cl2–MeOH (20:1, 15:1, 10:1, 9:1, 8:2, 7:3, 6:4, 1:1, 0:100, v/v) to yield nine fractions (L41–L49). L46 (183 mg) was further purified through semi-preparative HPLC (10–70% MeCN, 60 min, flow rate 15 mL/min) to yield 11 (tR of 31.35 min, 6.1 mg). L47 and L48 were combined and named as L47. The fraction L47 (267 mg) underwent purification via semi-preparative HPLC (15–75% MeCN, 60 min, flow rate 15 mL/min), which yielded nine sequentially eluted fractions (L471–L479) based on retention time. L476 (175 mg) was further purified through semi-preparative HPLC (45% MeCN, flow rate 15 mL/min) to yield 12 (tR of 17.14 min, 35.5 mg).
L5 (4.2 g) was purified by semi-preparative HPLC (10–37% MeCN, 80 min, flow rate 15 mL/min) to yield 3 (tR of 50.42 min, 28.6 mg), 6 (tR of 6.86 min, 33.8 mg), 7 (tR of 30.93 min, 358.6 mg), L52, L53, L58 and L510. L52 and L53 were combined and named as L52. L52 (40 mg) was further purified through semi-preparative HPLC (10% MeOH, flow rate 15 mL/min) to yield 5 (tR of 9.71 min, 5.4 mg). L58 (41.2 mg) was further purified by semi-preparative HPLC (37% MeOH, flow rate 15 mL/min) to yield 8 (tR of 33.75 min, 8.2 mg). L510 (139 mg) was further purified through semi-preparative HPLC (40% MeOH, flow rate 15 mL/min) to yield 9 (tR of 31.69 min, 85.9 mg).
L6 (1.8 g) was further separated by semi-preparative HPLC (5–75% MeOH, 60 min, flow rate 15 mL/min) to yield 1 (tR of 36.47 min, 54 mg), 4 (tR of 14.55 min, 552.1 mg), 10 (tR of 32.62 min, 28 mg) and L68. L68 (13 mg) was further purified by preparative HPLC with a DAICEL AD column (n-Hexane: EtOH = 9: 1, flow rate 1 mL/min) to yield 2 (tR of 9.32 min, 4 mg).
3.4 Compound characterization
Panunoid A (1). White amorphous powder. [α]D20 − 131.8 (c 0.13, MeOH); 1H and 13C NMR spectroscopic data see Table 1; HR-ESI–MS m/z 221.1151 [M + Na]+ (calculated for C11H18O3Na+, m/z 221.1154).
panunoid B (2). White amorphous powder. [α]D20 − 227.2 (c 0.14, MeOH); 1H and 13C NMR spectroscopic data see Table 1; HR-ESI–MS m/z 199.1326 [M + H]+ (calculated for C11H19O3+, m/z 199.1334).
panunoid C (3). Brown oil. [α]D20 − 108.8 (c 0.12, MeOH); 1H and 13C NMR spectroscopic data see Table 1; HR-ESI–MS m/z 234.0596 [M–H]– (calculated for C12H12NO2S–, m/z 234.0589), 218.0644 [M–H2O + H]+ (calculated for C12H12NOS+, m/z 218.0640).
panunoid D (4). Colorless crystal. [α]D20 − 255.7 (c 0.11, MeOH); 1H and 13C NMR spectroscopic data see Table 1. Crystal data for 4: Empirical formula C11H18O5; formula weight = 230.25; Temperature/K = 193.00; monoclinic space group C2; unit cell dimensions a = 38.2965(9) Å, b = 6.6639(2) Å, c = 9.2619(2) Å, α = γ = 90°, β = 98.7670(10)°; V = 2336.06(10) Å3; Z = 8; ρcalc = 1.309 g/cm3; μ = 0.863 mm−1; F(000) = 992.0; crystal size 0.15 × 0.14 × 0.12 mm3; radiation CuKα (λ = 1.54178); 2θ range for data collection 9.662 to 136.784°; index ranges − 46 ≤ h ≤ 46, − 8 ≤ k ≤ 7, − 11 ≤ l ≤ 11; reflections collected 38,710; independent reflections 4263 [Rint = 0.0365, Rsigma = 0.0196]; data/restraints/parameters 4263/1/303; goodness-of-fit on F2 = 1.042; final R indexes [I ≥ 2σ (I)]: R1 = 0.0283, wR2 = 0.0723; final R indexes [all data]: R1 = 0.0290, wR2 = 0.0730; largest diff. peak/hole 0.17/-0.20 e Å−3; Flack parameter 0.03(4). Crystallographic data for compound 4 have been deposited at the Cambridge Structural Database with the number of CCDC-2417279.
7-desoxyphenepoxydol (7). White crystal. 1H and 13C NMR spectroscopic data see Table 2. HR-ESI–MS m/z 197.1174 [M + H] + (calculated for C11H17O3+, m/z 197.1178). Crystal data for 7: Empirical formula C11H16O3; formula weight = 196.24; Temperature/K = 100.00(11); tetragonal space group P43; unit cell dimensions a = 14.6405(2) Å, b = 14.6405(2) Å, c = 4.81930(10) Å, α = β = γ = 90°; V = 1032.99(4) Å3; Z = 4; ρcalc = 1.262 g/cm3; μ = 0.740 mm−1; F(000) = 424.0; crystal size 0.35 × 0.06 × 0.05 mm3; radiation CuKα (λ = 1.54184); 2θ range for data collection 6.036 to 147.882°; index ranges − 16 ≤ h ≤ 18, − 14 ≤ k ≤ 18, − 5 ≤ l ≤ 5; reflections collected 7787; independent reflections 2024 [Rint = 0.0352, Rsigma = 0.0270]; data/restraints/parameters 2024/1/131; goodness-of-fit on F2 = 1.100; final R indexes [I ≥ 2σ (I)]: R1 = 0.0384, wR2 = 0.1022; final R indexes [all data]: R1 = 0.0393, wR2 = 0.1026; largest diff. peak/hole 0.14/-0.20 e Å−3; Flack parameter 0.04(13). Crystallographic data for compound 7 have been deposited at the Cambridge Structural Database with the number of CCDC-2496230.
(1S,3S,4R,6S,7S)-3,12-Dihydroxycadin-10(15)-ene (12). Colorless crystal. 1H NMR (400 MHz, CD3OD): δ 4.60 (d, J = 1.8 Hz, 1H, H-15), 4.50 (brs, 1H, H-15), 3.82 (q, J = 2.9 Hz, 1H, H-3), 1.18 (s, 3H, H-13), 1.12 (s, 3H, H-14), 0.95 (d, J = 6.9 Hz, 3H, H-11). 13C NMR (101 MHz, CD3OD): δ 154.5 (C-10), 104.0 (C-15), 74.7 (C-12), 71.1 (C-3), 54.0 (C-7), 48.1 (C-1), 40.1 (C-6), 38.1 (C-2), 38.0 (C-9), 37.6 (C-4), 35.8 (C-5), 32.4 (C-8), 31.5 (C-14), 24.8 (C-13), 19.4 (C-11). Crystal data for 12: Empirical formula C120H208O16; formula weight = 1906.85; Temperature/K = 149.99(10); triclinic space group P1; unit cell dimensions a = 13.31910(10) Å, b = 14.7838(2) Å, c = 17.4435(2) Å, α = 114.1670(10)°, β = 92.5210(10)°, γ = 105.8640(10)°; V = 2966.42(6) Å3; Z = 1; ρcalc = 1.067 g/cm3; μ = 0.532 mm− 1; F(000) = 1056.0; crystal size 0.2 × 0.13 × 0.12 mm3; radiation CuKα (λ = 1.54184); 2θ range for data collection 5.642 to 147.832°; index ranges − 16 ≤ h ≤ 16, − 17 ≤ k ≤ 18, − 21 ≤ l ≤ 21; reflections collected 74,242; independent reflections 22,440 [Rint = 0.0331, Rsigma = 0.0318]; data/restraints/parameters 22,440/3/1263; goodness-of-fit on F2 = 1.042; final R indexes [I ≥ 2σ (I)]: R1 = 0.0340, wR2 = 0.0857; final R indexes [all data]: R1 = 0.0382, wR2 = 0.0875; largest diff. peak/hole 0.31/-0.21 e Å−3; Flack parameter 0.04(4). Crystallographic data for compound 12 have been deposited at the Cambridge Structural Database with the number of CCDC-2498699.
3.5 Cytotoxicity assay
Two cancer cell lines, human lung cancer A-549 cells and human liver cancer HepG2 cells were used in the cytotoxic assay. Cells were digested with 0.25% trypsin and collected, followed by centrifugation at 1000 rpm for 4 min. After discarding the supernatant, 4 mL of fresh medium was added to resuspend the cell pellet. A 20 μL aliquot of cell suspension was mixed with 20 μL trypan blue solution for cell counting, with cell viability required to exceed 90%. The cell density was adjusted to 2000 cells/100 μL. A 96-well plate was loaded with 100 μL cell suspension per well and incubated overnight at 37℃ with 5% CO2 to allow cell adhesion. Test compounds at specified concentrations were added according to experimental requirements, with blank wells (cell-free wells) established as background controls. After 24, 48, and 72 h of treatment respectively, the culture medium was removed and replaced with 100 μL fresh medium containing 0.5% FBS per well. Subsequently, 10 μL CCK-8 solution was added to each well, followed by 1-h incubation at 37 ℃ (avoiding bubble formation that may interfere with OD readings). The optical density was measured at 450 nm using a microplate reader, and calculate the cell viability using Eq. (1). The IC50 values of compounds were calculated by Graphpad Prism 10.1.2 (GraphPad Software Inc., San Diego, CA, USA). All determinations were carried out in triplicate.
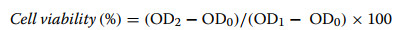
|
(1) |
where OD0 is the noise group, OD1 is the blank control group, and OD2 is the test group.
3.6 ECD calculation
The calculated structures were optimized with the Gaussian 09 package [35]. The B3LYP functional was used to all calculations in combination with the D3BJ dispersion correction. Time-dependent density functional theory (TD-DFT) were used to calculated the energy levels of the excited singlets. ECD spectrum were taken with multiwfn 3.8 software. The spectral data of different conformations were weighted to generate conformational average spectra.
4 Conclusions
In summary, this study focused on the rice fermentation products of P. rudis. A series of secondary metabolites were systematically characterized by a combination of multiple separation and characterization methods. Among them, four new compounds were found and the absolute configurations of 7 and 12 were determined to be unreported. At the same time, a reasonable biosynthetic pathway for 1–7 was inferred. From the results, it can be seen that compared with other substrates, solid-state fermentation using rice as the matrix tends to produce metabolites with carbonyl groups reduced to hydroxyl groups. In addition, compounds with potential cytotoxicity were screened. The results showed that 1, 3–6, 8, 9, and 12 exhibited cytotoxicity on A-549 and HepG2 cells, and 8 and 12 exhibited dose-dependent inhibitory effects on the two cell lines. This study not only expands the chemical profile of P. rudis solid-state fermentation products, but also reveals that the integration of the fungal enzymatic machinery with nutrient-rich substrates could provide direction for discovering metabolites with both structural diversity and bioactivity.
Notes
Acknowledgements
This work was supported by Major Program of Natural Science Foundation of the Higher Education Institutions of Jiangsu Province of China (No. 24KJA360001) and Open Project of Jiangsu Provincial Key Laboratory of Palygorskite Science and Applied Technology (No. HPZ202402).
Author contributions
Yun Liu: Writing—original draft, Methodology, Data curation, Investigation. Jian-qiang Zhao: Writing—review and editing, Data curation, Investigation. Yan-long Yang: Methodology, Investigation. Han-bing Yuan: Writing—original draft. Yan-ming Wang: Writing—review and editing, Methodology, Investigation. Jun Yuan: Writing—review and editing, Funding acquisition. The author(s) read and approved the final manuscript.
Data availability
Data will be made available on request.
Declarations
Competing interests
The authors declare no competing financial interest.
References
-
1.Wei F, Hong YZ, Liu JJ, et al. Gongronella sp induces overproduction of laccase in Panus rudis. J Basic Microbiol 2010;50(1): 98-103. CrossRef PubMed Google Scholar
-
2.Zhang M, Wu F, Wei ZY, et al. Characterization and decolorization ability of a laccase from Panus rudis. Enzyme Microb Technol 2006;39(1): 92-7. CrossRef PubMed Google Scholar
-
3.Yue L, Chen JL, Tuo YL, et al. Taxonomy and phylogeny of Panus (Polyporales, Panaceae) in China and its relationship with allies. MycoKeys 2024;105: 267-94. CrossRef PubMed Google Scholar
-
4.Gressler M, Löhr NA, Schäfer T, et al. Mind the mushroom: natural product biosynthetic genes and enzymes of Basidiomycota. Nat Prod Rep 2021;38(4): 702-22. CrossRef PubMed Google Scholar
-
5.Yang YL, Zhou M, Yang L, et al. A mushroom P450-monooxygenase enables regio- and stereoselective biocatalytic synthesis of epoxycyclohexenones. Angew Chem Int Ed 2023;62(49): 202313817. CrossRef PubMed Google Scholar
-
6.Song JG, Ha LS, Ki DW, et al. Chemical constituents of the culture broth of Panus rudis. Mycobiology 2021;49(6): 604-6. CrossRef PubMed Google Scholar
-
7.Lee YY, Ullah HMA, Ha LS, et al. Isopanepoxydone inhibits oxidative damage in murine alveolar macrophages via NRF2 and NLRP3 inflammasome. Immunopharmacol Immunotoxicol 2022;44(3): 347-54. CrossRef PubMed Google Scholar
-
8.Yu HP, Hao XJ, Gao YG, et al. Precursor-directed biosynthesis of panepoxydone derivatives with nitric oxide production inhibitory activity. ChemBioChem 2025;26(1): 202400691. CrossRef PubMed Google Scholar
-
9.Porco JA, Su S, Lei XG, et al. Total synthesis and structure assignment of (+)-hexacyclinol. Angew Chem Int Ed Engl 2006;45(35): 5790-2. CrossRef PubMed Google Scholar
-
10.Rychnovsky SD. Predicting NMR spectra by computational methods: structure revision of hexacyclinol. Org Lett 2006;8(13): 2895-8. CrossRef PubMed Google Scholar
-
11.Williams AJ, Elyashberg ME, Blinov KA, et al. Applying computer-assisted structure elucidation algorithms for the purpose of structure validation: revisiting the NMR assignments of hexacyclinol. J Nat Prod 2008;71(4): 581-8. CrossRef PubMed Google Scholar
-
12.Li L, Wang YZ, Chen NX, et al. Exploring diversity through dimerization in natural products by a rational tandem mass-based molecular network strategy. Org Lett 2023;25(22): 4016-21. CrossRef PubMed Google Scholar
-
13.Ding JH, Li ZH, Feng T, et al. A new cadinane sesquiterpenoid from cultures of the Basidiomycete Panus conchatus. Nat Prod Res 2018;32(19): 2333-7. CrossRef PubMed Google Scholar
-
14.Wang SX, Chen BS, Zhang ZJ, et al. Isolation, structural elucidation and biosynthetic pathway of bioactive prenyl quinone compounds from Panus lecomtei based on untargeted metabolomics combined with molecular networking. Food Chem 2025;463: 9. CrossRef PubMed Google Scholar
-
15.Wang SX, Zhao RL, Guo C, et al. New neroterpenoid compounds from the culture of mushroom Panus lecomtei. Chin J Nat Med 2020;18(4): 268-72. CrossRef PubMed Google Scholar
-
16.Palasarn S, Tanyapanyachon P, Vichai V, et al. Chromene dimers from cultures of Basidiomycete Panus similis. J Nat Prod 2025;88(3): 777-84. CrossRef PubMed Google Scholar
-
17.Gong ZH, Wu ZY, Yang Q, et al. Influences of lactic acid bacteria strains on the flavor profiles, metabolites and quality characteristics of red yeast rice produced by solid-state fermentation. Food Res Int 2024;197: 115172. CrossRef PubMed Google Scholar
-
18.He JY, Li M, Gao MX, et al. Differential volatile compounds between rice and tartary buckwheat by solid-state fermentation with Monascus purpureus. Int J Food Microbiol 2025;435: 111181. CrossRef PubMed Google Scholar
-
19.Zhang BB, Xing HB, Jiang BJ, et al. Using millet as substrate for efficient production of monacolin K by solid-state fermentation of Monascus ruber. J Biosci Bioeng 2018;125(3): 333-8. CrossRef PubMed Google Scholar
-
20.Zhu JS, Lu F, Liu DD, et al. The process of solid-state fermentation of soybean meal: antimicrobial activity, fermentation heat generation and nitrogen solubility index. J Sci Food Agric 2024;104(6): 3228-34. CrossRef PubMed Google Scholar
-
21.Peng WW, Huang Q, Ke X, et al. Koningipyridines A and B, two nitrogen-containing polyketides from the fungus Trichoderma koningiopsis SC-5. Nat Prod Bioprospect 2024;14(1): 13. CrossRef PubMed Google Scholar
-
22.Wu WY, Wei X, Liao Q, et al. Structurally diverse polyketides and alkaloids produced by a plant-derived fungus Penicillium canescens L1. Nat Prod Bioprospect 2025;15(1): 12. CrossRef PubMed Google Scholar
-
23.Yin Q, Han JY, Yang GX, et al. New sesquiterpenoids with anti-inflammatory effects from phytopathogenic fungus Bipolaris sorokiniana 11134. Nat Prod Bioprospect 2025;15(1): 13. CrossRef PubMed Google Scholar
-
24.Liu S, Sun CZ, Ha Y, et al. Novel antibacterial alkaloids from the Mariana Trench-derived actinomycete Streptomyces sp. SY2255. Tetrahedron Lett 2024;137: 154935. CrossRef PubMed Google Scholar
-
25.Ichihara A, Kimura R, Sakamura S. Synthesis of 7-desoxypanepoxydol. Agric Biol Chem 1975;39(2): 555-6. CrossRef PubMed Google Scholar
-
26.Shotwell JB, Koh B, Choi HW, et al. Inhibitors of NF-κB signaling: design and synthesis of a biotinylated isopanepoxydone affinity reagent. Bioorg Med Chem Lett 2002;12(23): 3463-6. CrossRef PubMed Google Scholar
-
27.Vásquez R, Rios N, Solano G, et al. Lentinoids A-D, new natural products isolated from Lentinus strigellus. Molecules 2018;23(4): 773. CrossRef PubMed Google Scholar
-
28.Wang QL, She XG, Ren XF, et al. The first asymmetric total synthesis of several 3,4-dihydroxy-2,2-dimethyl-chroman derivatives. Tetrahedron Asymmetry 2004;15(1): 29-34. CrossRef PubMed Google Scholar
-
29.Zheng YB, Zhao BB, Lu CH, et al. Isolation, structure elucidation and apoptosis-inducing activity of new compounds from the edible fungus Lentinus striguellus. Nat Prod Commun 2009;4(4): 501-6. PubMed Google Scholar
-
30.Fan XL, Cao YG, Zeng MN, et al. Six new compounds from the herbaceous stems of Ephedra intermedia Schrenket C. A. meyer and their lung-protective activity. Molecules 2024;29(2): 432. CrossRef PubMed Google Scholar
-
31.Collins DO, Ruddock PLD, de Grassea JC, et al. Microbial transformation of cadina-4,10(15)-dien-3-one, aromadendr-1(10)-en-9-one and methyl ursolate by Mucor plumbeus ATCC 4740. Phytochemistry 2002;59(5): 479-88. CrossRef PubMed Google Scholar
-
32.Husain SM, Schätzle MA, Lüdeke S, et al. Unprecedented role of hydronaphthoquinone tautomers in biosynthesis. Angew Chem Int Ed 2014;53(37): 9806-11. CrossRef PubMed Google Scholar
-
33.Zhang R, He JW, Dong ZW, et al. Genomic and experimental data provide new insights into luciferin biosynthesis and bioluminescence evolution in fireflies. Sci Rep 2020;10(1): 15882. CrossRef PubMed Google Scholar
-
34.Gao Y, Yang J, Yang XL, et al. Novel dibenzofuran and biphenyl phytoalexins from Sorbus pohuashanensis suspension cell and their antimicrobial activities. Fitoterapia 2021;152: 104914. CrossRef PubMed Google Scholar
-
35.Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09, Revision D. 01. Gaussian: Wallingford CT, 2009. PubMed Google Scholar
Copyright information
© The Author(s) 2026
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
