
First discovery of triterpenoids and sterols from Cotinus coggygria var. cinereus Engl. with anti-inflammatory and antibacterial activities
Abstract
This study led to the isolation of 17 triterpenoids, including two lanostane-type (1 and 2), two dammarane-type (3 and 7), ten tirucallane-type (4 and 8−16), and three oleanane-type (17−19) triterpenoids, as well as nine sterols (5, 6, and 20−26) from Cotinus coggygria var. cinereus Engl, which were first discovered from the genus Cotinus. Among them, coggygrenoids A−D (1−4), coggygrerol A (5), and coggygrerol B (6) are undescribed compounds. Additionally, seven flavonoids (27−33) were also isolated from this plant. Compound 15 displayed inhibitory activities in LPS-induced RAW 264.7 cells with an IC50 value 6.81 ± 0.15 μM. Molecular docking demonstrated that 15 exhibited favorable affinity for NLRP3 and iNOS. In vitro and in vivo antibacterial evaluations indicated that coggygrnoid C (3) exhibited significant inhibitory activity against methicillin-resistant Staphylococcus aureus ATCC BAA-1717 (USA300), with An MIC of 8 μg/mL. Further mechanistic investigations demonstrated that 3 exerted antibacterial activity by compromising the integrity of the cell wall and membrane. Notably, the combination of 3 with ampicillin exhibited an additive antibacterial effect. In the Galleria mellonella infection model, compound 3 exhibited comparable activity to that of the positive control at 20 mg/kg. These findings suggest that triterpenoids of C. coggygria are potential antibacterial lead agents.Graphical Abstract
Keywords
Cotinus coggygria var. cinereus Engl. Triterpenoids Sterols Anti-inflammatory activity Antibacterial activity1 Introduction
Cotinus plants belong to the Anacardiaceae family and are categorized as deciduous shrubs or small trees [1]. These species are regarded as excellent ornamental tree species for landscaping purposes and served as pioneer tree species for afforestation on barren hills. Cotinus is rich in resources and widely distributed in Southern Europe, North America, China, and other regions. In China, Cotinus is primarily distributed in the mid-to-low altitude mountainous areas of Shaanxi, Shanxi, Henan, and Hebei provinces, with the main species being Cotinus coggygria var. cinereus Engl. [2]. C. coggygria contains a variety of chemical components, including flavonoids [3], gallic acid [4], and essential oils [5]. Historically, it has been extensively employed in traditional Chinese medicine and used for treating various diseases, including jaundice-type hepatitis, burns from fire or water, and lacquer dermatitis. Previous studies have shown that the extracts and chemical constituents of C. coggygria display extensive pharmacological effects, particularly in antioxidant [6, 7] hypotensive [8], liver damage repair [9], and antibacterial effects [10]. Triterpenoids are natural products composed of a basic nucleus consisting of six isoprene units totalling 30 carbon atoms, which have complex And diverse structures, encompassing over 100 different skeletal frameworks. A few of these are linear, monocyclic, bicyclic or tricyclic. The most common types are tetracyclic and pentacyclic triterpenes. Tetracyclic triterpenes are primarily classified as lanostane-, tirucallane-, dammarane- and cucurbitane-type. As important plant secondary metabolites, triterpenoids are involved in various physiological processes, such as oxidative stress, and exhibit pharmacological effects, including anti-inflammatory, neuroprotective, anti-Alzheimer's disease, antifungal and antitumour activities. As far as we know, no studies have been reported on the triterpenoids along with their biological activities in C. coggygria.
As part of an ongoing program of research into the identification of novel bioactive substances from traditional medicinal plants in the Tsinling Mountains [11-17], a phytochemical investigation was conducted on the tree stems and bark of C. coggygria. This investigation resulted in the isolation of a series of triterpenoids and sterols from the plant for the first time. The structures of the previously undescribed triterpenoids and sterols were elucidated using HRESIMS, NMR spectroscopy, and electronic circular dichroism (ECD) calculations. Furthermore, all isolates were evaluated for their anti-inflammatory activity in lipopolysaccharide (LPS) stimulated nitric oxide (NO) production in RAW 264.7 cells and their antibacterial activity against five gram-positive and five gram-negative bacteria. Herein, we describe the isolation, structural elucidation, biological activities, and mechanism of action of the isolated triterpenoids and sterols.
2 Results and discussion
2.1 Structure elucidation
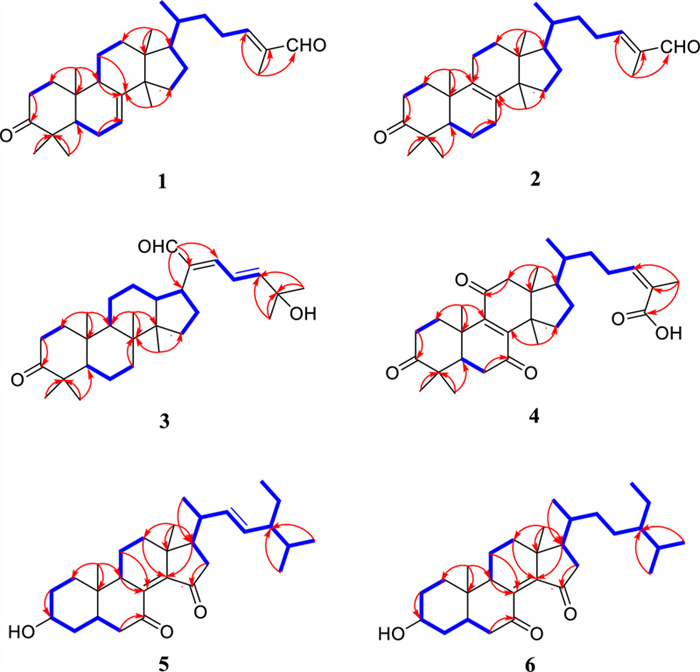
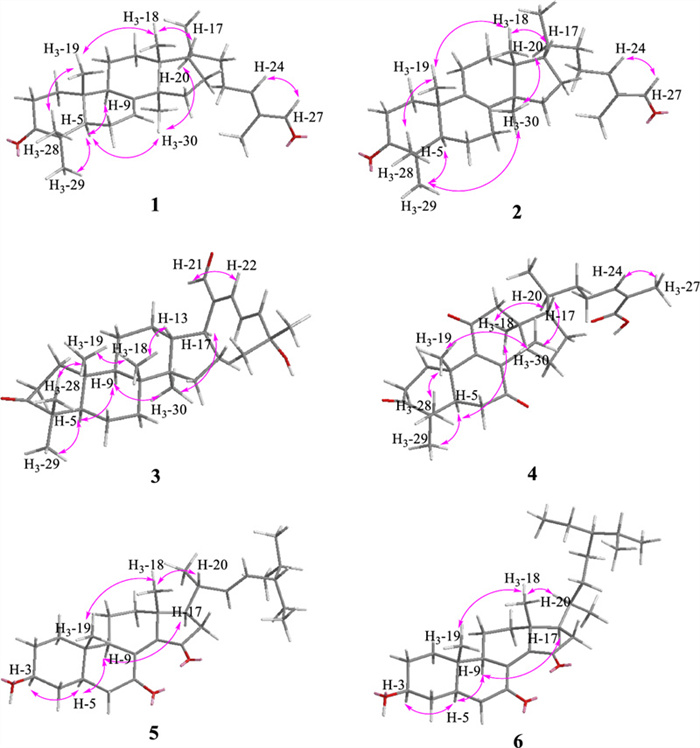
Coggygrnoid A (1) was obtained as a colorless solid with the molecular formula C30H46O2, as established by a [M + Na]+ ion peak at m/z 461.3382 in its HRESIMS spectrum (calcd for C30H46O2Na, 461.3390), implying 11 degrees of unsaturation. The 1H NMR data (Table 1) of 1 indicated the presence of one aldehyde proton [δH 9.39 (1H, s, H-27)] and two olefinic protons [δH 6.49 (1H, t, J = 7.4 Hz, H-24), 5.31 (1H, m, H-7)]. In the high field, six tertiary methyl groups [δH 1.75 (3H, s, H3-26), 1.11 (3H, s, H3-29), 1.04 (3H, s, H3-28), 1.02 (3H, s, H3-30), 1.00 (3H, s, H3-19), 0.81 (3H, s, H3-18)], and a secondary methyl group [0.91 (3H, d, J = 6.1 Hz, H3-21)] were detected. The 13C NMR and HSQC spectra of 1 (Table 1) showed thirty carbon signals sorted as one keto-carbonyl [δC 217.0 (C-3)], one aldehyde [δC 195.5 (C-27)], two double bonds [δC 118.2 (C-7), 145.7 (C-8), 155.4 (C-24), 139.2 (C-25)], four quaternary carbons [δC 48.0 (C-4), 35.1 (C-10), 43.6 (C-13), 51.4 (C-14)], four methines [δC 52.4 (C-5), 48.5 (C-9), 53.3 (C-17), 36.0 (C-20)], nine methylenes [δC 38.6 (C-1), 35.1 (C-2), 24.5 (C-6), 18.4 (C-11), 33.9 (C-12), 34.0 (C-15), 28.6 (C-16), 33.7 (C-22), 26.6 (C-23)], and seven methyls [δC 22.4 (C-18), 12.9 (C-19), 18.6 (C-21), 9.2 (C-26), 24.7 (C-28), 21.7 (C-29), 27.6 (C-30)]. Analysis of the above spectral data suggests that compound 1 is a lanostane triterpenoid with an α,β-unsaturated aldehyde structural fragment at the end of its side chain [18]. Further Analysis of the 2D NMR data of compound 1 led to the determination of its planar structure. The 1H-1H COSY correlations (Fig. 2) revealed the presence of four spin-coupling systems (H2-1/H2-2; H-5/H2-6/H-7; H-5/H2-11/H2-12; H2-15/H2-16/H-17/H-20/(H3-21)/H2-22/H2-23/H-24) in 1. The HMBC correlations (Fig. 2) of H3-28/H3-29 with C-3, C-4, and C-5; H3-19 with C-1, C-9, and C-10; H-2 with C-3; H-6 with C-8; H3-18 with C-12, C-13, and C-17; H3-30 with C-8, C-14, and C-15; and H3-26 with C-24, C-25, and C-27 established the carbon connectivity of 1, and the planar structure of compound 1 was then determined as (24E)-3-oxo-5α-lanosta-7,24-dien-27-al. The relative configuration of compound 1 was established by analysing its NOESY spectrum. The NOESY correlations (Fig. 3) observed between H3-19 and H3-18/H3-28, as well as between H3-18 and H-17 indicate a co-facial configuration, and assigned as β-orientation. Meanwhile, the NOESY correlations between H-5 and H-9/H3-29/H3-30 revealed that they were α-oriented. The double bond Δ24, 25 was determined as Z-configuration by the NOE-correlation of CHO-27 with H-24. To confirm the absolute structure of 1, experimental and calculated ECD spectra (Fig. 4) were obtained, which verified the structure of 1, as shown in Fig. 1.
1H NMR (400 MHz) and 13C NMR (100 MHz) data of 1–4 in CDCl3
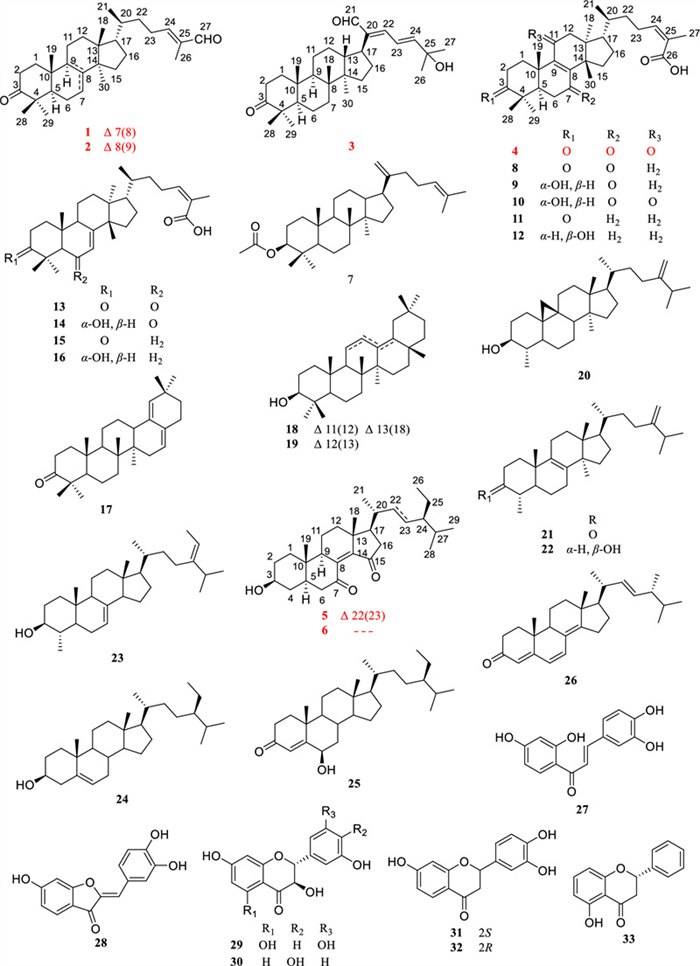
Structures of compounds 1–33
Key HMBC and 1H-1H COSY correlations of compounds 1–6
Key NOESY correlations of compounds 1–6
Coggygrnoid B (2) possessed a molecular formula of C30H46O2 (eight degrees of unsaturation), as determined by the quasi-molecular ion peak 461.3395 [M + Na]+ (calcd for C30H46O2Na, 461.3390) in its HRESIMS spectrum. The characteristic signals of one aldehyde proton at δH 9.39 (1H, s, H-27), an olefinic proton at δH 6.49 (1H, t, J = 7.2 Hz, H-24), and seven methyl signals at δH 1.75 (3H, s, H3-26), 1.09 (3H, s, H3-28), 1.05 (3H, s, H3-29), 1.05 (3H, s, H3-19), 0.90 (3H, d, J = 6.5 Hz, H3-21), 0.90 (3H, s, H3-30), And 0.76 (3H, s, H3-18) were presented in the 1H NMR spectrum (Table 1). A total of 30 carbon resonances were well resolved in the 13C NMR spectrum (Table 1), which were ascribed to seven methyls, ten methylenes, five methines (including two sp2 carbons), and eight quaternary carbons (one of which was carbonyl). Furthermore, a comparison of the 1H and 13C NMR data of 2 with those of compound 1 revealed close similarities. The Major difference was that the double bond at Δ 7(8) in 1 appeared at Δ 8(9) in 2, which was supported by the chemical shifts of C-8 (δC 134.8), and C-9 (δC 132.7) and key HMBC correlations (Fig. 2) from H2-7 to C-8 and from H2-11 to C-9. Based on the biosynthetic relationship, combined with the NOESY correlations (Fig. 3) of H3-18/H-17, H3-18/H3-19, H3-19/H3-28, H3-29/H-5, H3-30/H-5, and CHO-27/H-24, the absolute configuration of 2 was assigned to be the same as those of 1. Although we did not obtain the CD curve of 2, its specific optical rotation value, which was similar to that of 1, also confirmed this assignment. Thus, the structure of compound 2 was elucidated as (24E)-3-oxo-5α-lanosta-8,24-dien-27-al.
Coggygrnoid C (3) was obtained as a faint yellowish gum and showed the molecular formula C30H46O3 based on the HRESIMS ion at m/z 477.3330 [M + Na]+ (calcd for C30H46O3Na, 477.3339). The 13C NMR and HSQC spectra (Table 1) showed 30 carbon resonances, which were classified as seven quaternary carbons (one ketone carbonyl and one olefinic), eight methines (one aldehyde and three olefinic), eight methylenes, and seven methyls by their chemical shifts and hybridization states. Combining the above data with the unsaturation of compound 3 indicates that it contains four rings in its structure. Additionally, the presence of one conjugated diene group [δH 6.77 (1H, d, J = 13.9 Hz, H-22), 6.77 (1H, q, J = 13.9 Hz, H-23), 6.30 (1H, d, J = 13.9 Hz, H-24)], and seven tertiary methyls [δH 1.40 (3H, s, H3-26), 1.40 (3H, s, H3-27), 1.08 (3H, s, H3-28), 1.05 (3H, s, H3-19), 1.04 (3H, s, H3-29), 0.92 (3H, s, H3-18), And 0.92 (3H, s, H3-30)] were evident from the 1H NMR data (Table 1). The above data indicated that compound 3 was based on a dammarane triterpenoid skeleton similar to that of aglaiabbreviatin E (7) [19], with the main difference being the structure of the side chains. The exact structure of 3 was deduced from the 1H-1H COSY and HMBC spectra (Fig. 2). The HMBC correlations arising from CHO-21 to C-17, C-20, and C-22; the gem-dimethyl protons (H3-26 and H3-27) to C-24, and C-25, and the spin system (H-22/H-23/H-24) indicated the presence of an α,β,γ,δ,-unsaturated aldehyde, which is consistent with the UV absorption curve. Compound 3 shared the same relative configuration as those of other dammarane-type triterpenoids. The NOESY correlations (Fig. 3) of H3-18/H-13, H3-18/H3-19, and H3-19/H3-28 indicated co-facial orientation, which was assigned as β. Also, the NOESY correlations (Fig. 3) of H3-29/H-5, H-9/H-5, H-9/H3-30, and H3-30/H-17 suggested that these were α-oriented. Therefore, the structure of compound 3 was elucidated as (21E,23E)-3-oxo-25-hydroxydammar-21-al-20,23-diene.
Coggygrnoid D (4) gave a molecular formula of C30H42O5 as determined by an HRESIMS [M−H]− peak at 481.2957 (calcd for C30H41O5, 481.2959). The 1H NMR spectrum (Table 1) exhibited the presence of seven methyl signals, including one methyl doublet at δH 0.90 (d, J = 6.6 Hz), five tertiary methyls, and one singlet methyl at δH 1.92. In addition, one olefinic proton was observed at δH 6.07 (t, J = 7.1 Hz). The 13C NMR spectrum (Table 1) displayed 30 carbon resonances. These were classified by HSQC experiment as three ketone carbonyls, one carboxyl group, two double bonds, four sp3 quaternary carbons, three sp3 methines, eight sp3 methylenes, and seven methyls in the structure. Comparing the NMR data of 4 with 3,7-dioxo-8, 24Z-tirucalladien-26-oic acid (8) revealed that the structures were closely related. The only difference was the presence of an additional carbonyl group at C-11 (δC 202.3). The two methylene proton resonances at δH 2.68 And 2.49, which exhibit larger coupling constant (J = 16.4 Hz) and an HMBC correlation with δC 202.3 And 153.6 (Fig. 2), may be assigned to H2-12 in the α-position relative to one of the carbonyl groups (δC 202.3). Moreover, the large downfield chemical shift at C-8 (δC 150.3) and upfield chemical shift at C-9 (δC 153.6) also confirmed that the olefinic bond was assigned between C-7 carbonyl and C-11carbonyl in 4. The relative configuration of compound 4 was determined by the following correlation signals on the NOESY spectrum: H3-19 and H3-28, H3-30; H3-30 and H-7; H-5 and H3-18, H3-29; H-20 and H3-18; H-24 and H3-27. The high match of the experimental and calculated ECD spectra showed that the absolute configurations of 4 were 5R,10S,13S,14S,17S,20S (Fig. 4). Thus, the structure of 4 was confirmed as 3,7,11-trioxo-8,24Z-tirucalladien-26-oic acid.
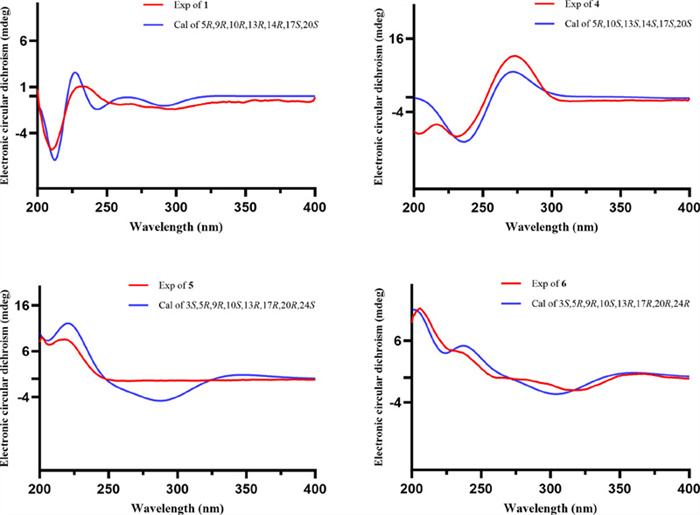
Experimental and calculated ECD curves of compounds 1 and 4–6
Coggygrerol A (5) was a yellowish gum. The molecular formula of 5 was deduced from the quasi-molecular ion peak [M + Na]+ at m/z 463.3179 (calcd for C29H44O3Na, 463.3183) in the HRESIMS spectrum. The 1H and 13C NMR data (Table 2) indicated the presence of two ketone carbonyls [δC 202.1 (C-7), 205.1 (C-15)], two double bonds [δC 143.0 (C-8), 148.4 (C-14), 136.2 (C-22), 131.6 (C-23); δH 5.14 (1H, m, H-22), 5.14 (1H, m, H-23)], one oxygenated methine [δC 70.5 (C-3); δH 3.65 (1H, m, H-3)], and six methyl group [δC 18.2 (C-18), 12.5 (C-19), 21.7 (C-21), 12.4 (C-26), 21.2 (C-28), 19.3 (C-29); δH 1.01 (3H, s, H3-18), 0.95 (3H, s, H3-19), 1.10 (3H, d, J = 7.2 Hz, H3-21), 0.79 (3H, d, J = 6.9 Hz, H3-26), 0.83 (3H, d, J = 6.4 Hz, H3-28), 0.79 (3H, d, J = 6.4 Hz, H3-29)]. The 1H and 13C NMR spectra of compound 5 (Table 2) exhibited a high degree of similarity to those of the previously reported compound (3S,8E,22E)-hydroxystigmast-8,22-diene-7,11-dione [20] and as a typical steroidal skeleton. According to the subsequent comprehensive comparison of the data obtained for the two compounds, the bi-conjugate with a trans double bond (C-8, C-9) and two ketone carbonyl groups (C-7, C-11) in (3S, 8E, 22E)-hydroxystigmast-8,22-diene-7,11-dione was assumed to have transformed to ∆8, 14 (double bond) and C-7/C-15 (two ketone carbonyl groups) in 5. This assumption was confirmed by the HMBC correlations from H2-6 to C-7; H-9, H2-11 to C-8; H2-12, H-17 to C-14; and those from H2-16 to C-15 (Fig. 2). The configuration of the ∆2, 3 double bond was deduced from the large coupling constant between the olefinic protons (δH 5.14, 1H, dd, J = 16.0, 8.2 Hz; δH 5.14, 1H, dd, J = 16.0, 8.2 Hz). The observed NOESY correlations between H-3/H-5, H-5/H-9, and H-9/H-17, determined that H-5, H-9, and H-17 were all located on the α-face of the steroid. Furthermore, the correlations between H3-18/H3-19 and H3-18/H-20 confirmed the β-orientation of C-18, C-19, and C-20 (Fig. 3). Based on the above evidence, the structure of 5 was unambiguously determined to be (3S, 8E, 22E)-hydroxystigmast-8,22-diene-7,15-dione.
1H NMR(400 MHz) and 13C NMR (100 MHz) data of 5 and 6 in CDCl3
Coggygrerol B (6) was a yellowish gum, and its HRESIMS showed m/z 465.3334 [M + Na]+, indicating a molecular formula of C29H46O3. By comparing the spectroscopic data (Table 2) of 6 with those of 5, it was found that 6 had a structure similar to that of 5, except that the double bond ∆22,23 was absent in 6. The main difference in the 1H NMR spectrum was the absence of double bonds at δH 5.14 (2H), which was supported by the 13C NMR spectrum. These changes were verified via the 1H-1H COSY correlations (Fig. 2) of H-20/H2-22/H2-23/H-24, together with the HMBC relationships (Fig. 2) of H-20, H3-21 with C-22, and H-24, H2-25 with C-23. Thus, the structure of 6 was determined as (3S,8E)-hydroxystigmast-8-ene-7,15-dione.
By comparing spectroscopic data with those reported previously, the 27 known compounds isolated from this plant were shown to be aglaiabbreviatin E (7) [19], 3,7-dioxo-8,24Z-tirucalladien-26-oic acid (8) [21], 3-hydroxy-7-oxo-8,24Z-tirucalladien-26-oic acid (9) [21], 7,11-dioxo-3-hydroxy-8,24Z-tirucalladien-26-oic acid (10) [21], isomasticadienoic acid (11) [22], 24Z-isomasticadienolic acid (12) [23], (9R)-3,6-dioxo-tirucalla-7, 24Z-dien-26-oic acid (13) [24], 3-hydroxy-6-oxo-7,24Z-tirucalladien-26-oic acid (14) [21], masticadienonic acid (15) [23], schinol (16) [21], 28-norolean-16,18-dien-3-one (17) [25], oleana-11:l3(18)-dien-3β-ol, (18) [26], β-amyrin (19) [27], cycloeucalenol (20) [28], obtusifolione (21) [29], obtusifoliol (22) [30], citrostadienol (23) [31], β-sitosterol (24) [32], 6-hydroxystigmast-4-en-3-one (25) [33], ergosta-4,6,8(14),22-tetraen-3-one (26) [34], butein (27) [35], sulfuretin (28) [36], 3,3',5',5,7-pentahydroflavanone (29) [37], fustin (30) [38], (−)-butin (31) [36], (+)-butin (32) [35], And 5-hydroxyflavanone (33) [39].
2.2 Effect of compounds on the LPS induced production of NO
Various diseases are accompanied by inflammation, and identifying small-molecule natural products with anti-inflammatory activity is a promising strategy for developing novel and effective therapeutic agents. Thus, the inhibitory effects of compounds 1−27 on LPS induced NO production in RAW 264.7 cells were assessed. Among them, compounds 5, 13, 15, 21, and 26 exhibited inhibitory activities in LPS induced RAW 264.7 cells, with IC50 values ranging from 6.81 ± 0.15 to 21.54 ± 0.35 μM (Table 3). Notably, compound 15 exhibited activity comparable to that of the positive control dexamethasone (IC50: 8.25 ± 0.6 μM). Molecular docking analysis revealed that compound 15 has a high affinity for NLRP3 and iNOS, with binding energies of −11.7 and −10.3 kcal/mol, respectively (Fig. 5). The above findings confirms that 15 exerts its anti-inflammatory effect by acting on NLRP3 and iNOS, revealing that NLRP3 and iNOS may be potential targets for the inhibitory activities of compound 15 in LPS-induced RAW 264.7 cells. These findings suggested that Coggygrerol A (5), (9R)-3,6-dioxo-tirucalla-7,24Z-dien-26-oic acid (13), masticadienonic acid (15), obtusifolione (21) and ergosta-4,6,8(14),22-tetraen-3-one (26) exhibited notable anti-inflammatory effects.
Inhibitory effects of compounds on NO production induced by LPS in RAW 264.7 cells
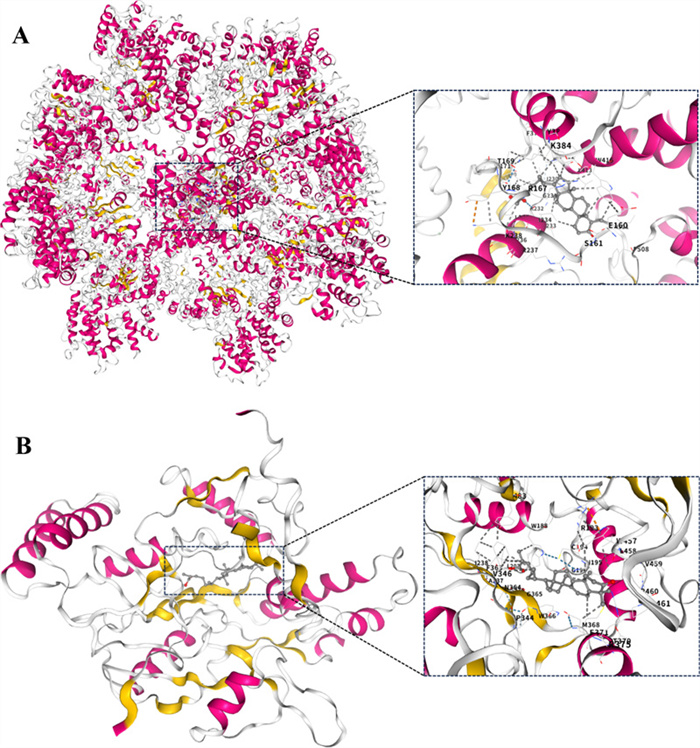
The molecular interactions of compound 15 with NLRP3 (A) and iNOS (B) by molecular docking simulation
2.3 Antibacterial activity of the isolated compounds in vitro
The inhibitory effects of the 27 triterpenoid And sterol compounds against the 10 bacterial strains were evaluated, and the results are presented in Table 4. Compounds 3, 4, 10, 13, 14, and 26 exhibited varying inhibitory effects against the tested bacteria, while the other compounds showed no significant inhibitory effects. Notably, all six compounds contain multiple carbonyl groups and conjugated systems, which are thought to be related to their antibacterial activities. Specifically, compound 3 demonstrated noteworthy antibacterial activity against MRSA ATCC BAA-1717(USA300) And MRSA 12228, exhibiting a MIC value of 8 μg/mL.
Antibacterial activity of compounds against test bacteria
2.4 Inhibitory effect of compound 3 on the growth of MRSA ATCC BAA-1717(USA300)
The growth curves for MRSA ATCC BAA-1717(USA300) treated with compound 3 are shown in Fig. 6A. In the negative control group, the OD600 first increased rapidly And then remained stable until the end of the experiment. Compared to the control, 0.25 × MIC And 0.5 × MIC of 3 against MRSA ATCC BAA-1717(USA300) slightly delayed the exponential phase of the bacterial cells. The logarithmic period of the cells was significantly delayed when a higher concentration of compound 3 (1 × MIC) was used. In addition, at 2 × MIC of 3 against MRSA171, the growth curve remained almost horizontal from beginning to end (i.e., no logarithmic period occurred), suggesting that the growth of the cells was completely inhibited (Fig. 6A). Consequently, the growth curve analysis revealed that compound 3 exhibited potent dose- and time-dependent inhibitory effects against MRSA ATCC BAA-1717(USA300).
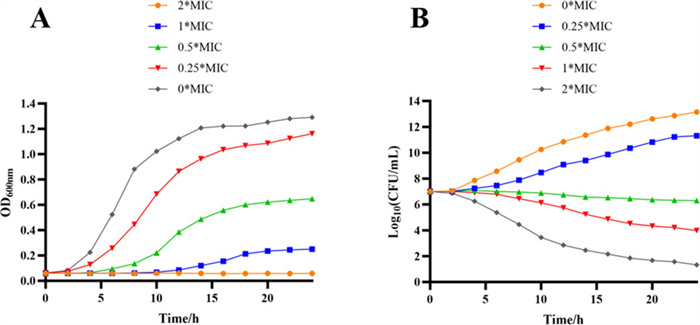
Growth curves (A) and Time-kill curves (B) of compound 3 against MRSA ATCC BAA-1717 (USA300)
2.5 Killing kinetics evaluation of compound 3 against MRSA ATCC BAA-1717(USA300)
In addition to the inhibition of bacterial growth, the rapid killing of bacteria is also a significant indicator of the antimicrobial activity of compounds. Consequently, compound 3 was selected for further evaluation of its bactericidal performance using a standard time-kill assay against MRSA ATCC BAA-1717(USA300). As illustrated in Fig. 6B, compound 3 rapidly eliminated MRSA ATCC BAA-1717(USA300) at 1 × MIC And 2× MIC.
2.6 Extracellular alkaline phosphatase (AKP) activity
The assessment of extracellular AKP levels can serve as a rapid indicator of cell wall damage and functional disruption. In this study, extracellular AKP levels were measured to evaluate the extent of cell wall damage caused by 3. As shown in Fig. 7, the AKP level increased with the concentration of 3. Compared to the blank control group, the AKP levels in the bacterial samples increased significantly. These findings suggest that 3 disrupts the cell wall, leading to AKP release.
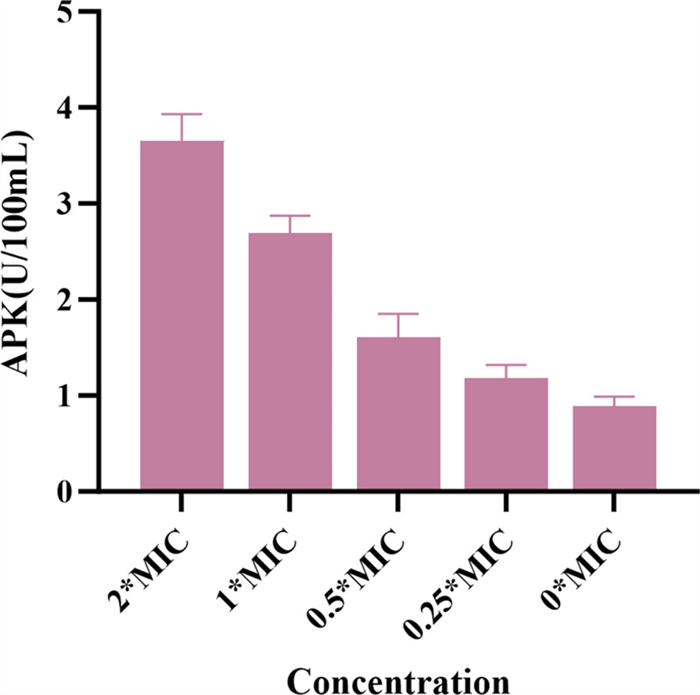
The effect of compound 3 on AKP activity in MRSA ATCC BAA-1717 (USA300)
2.7 Antibiofilm activity
The effect of 3 on the biofilm formation capacity of MRSA ATCC BAA-1717(USA300) was evaluated using a crystal violet staining assay. Compound 3 exhibited a significant inhibitory effect on the biofilm formation of MRSA ATCC BAA-1717 (USA300). When treated with 3 at 2 × MIC, biofilm formation was inhibited by 70.5%. Treatment with 3 at 1 × and 1/2 × MIC inhibited biofilm formation by 59.4% And 25.5%, respectively (Fig. 8).
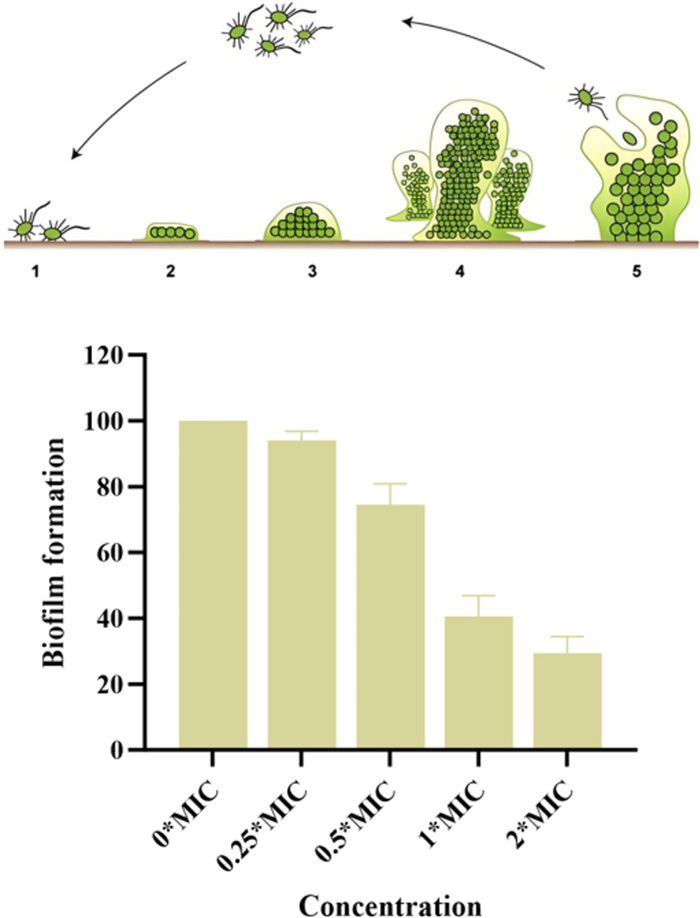
The effect of compound 3 on biofilm formation in MRSA ATCC BAA-1717 (USA300)
2.8 SEM analysis of MRSA ATCC BAA-1717(USA300)
To further demonstrate the effects of 3 against MRSA ATCC BAA-1717(USA300), morphological alterations in the bacteria after treatment with 3 were examined using a Scanning Electron Microscope (SEM), and the resulting images are depicted in Fig. 9. Untreated MRSA ATCC BAA-1717(USA300) cells exhibited intact and smooth cell surfaces (Fig. 9A), whereas MRSA ATCC BAA-1717(USA300) cells treated with compound 3 at 4 × the MIC exhibited severe damage and breakage (Fig. 9B). According to these SEM results, it was inferred that the possible antibacterial mechanism of 3 against MRSA ATCC BAA-1717(USA300) might involve the induction of abnormal morphology in the cell membranes, causing an increase in membrane permeability.
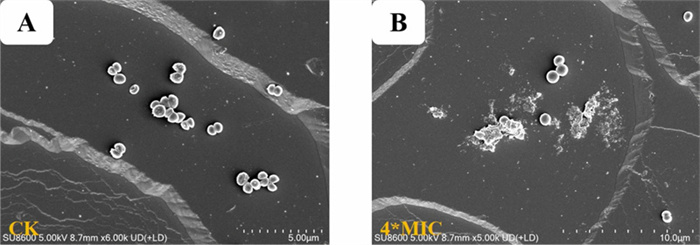
Scanning electron microscopy (SEM) images of MRSA ATCC BAA-1717 (USA300) treated with compound 3. A Control for MRSA ATCC BAA-1717 (USA300). B Treated by compounds 3
2.9 Combination susceptibility testing
Combination susceptibility testing revealed that 3 and ampicillin exerted an additive effect against MRSA ATCC BAA-1717(USA300) (Fig. 10). This combination targets multiple bacterial sites, thereby reducing the likelihood of simultaneous resistance development to both mechanisms. Moreover, the use of this combination may enable dose reduction of each individual agent, potentially minimizing toxicity and delaying the emergence of resistance.
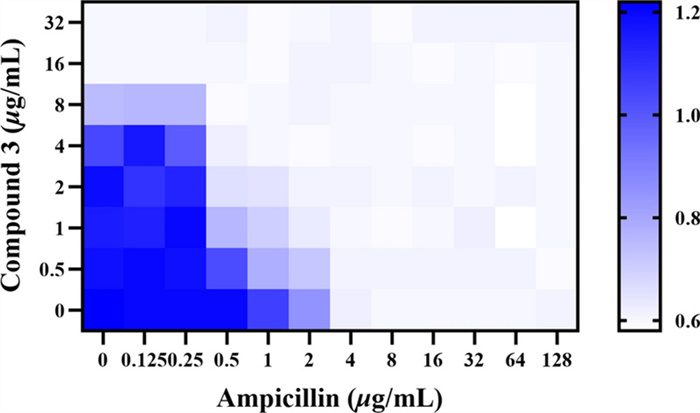
Antibacterial effect of the combination of compound 3 and ampicillin against MRSA ATCC BAA-1717 (USA300)
2.10 Antibacterial activity of compound 3 in vivo in the G. mellonella infection model
Based on the excellent antibacterial activity of compound 3, it was further evaluated in vivo using a G. mellonella infection model with vancomycin as the positive control, as in the in vitro experiments. As shown in Fig. 11, treatment with 3 significantly increased the survival rate and reduced the bacterial content in the body in a dose-dependent Manner from 5 to 20 mg/kg. Moreover, at the high dose of 20 mg/kg, the treatment rate was 80%, consistent with that of the positive control group. These findings suggest that compound 3 has potential as a candidate for future antibiotic development.
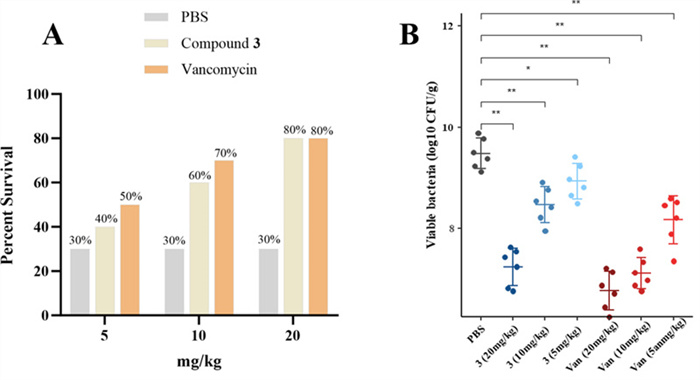
In vivo antibacterial activity of compound 3. A Survival rate of infected G. mellonella after treatment with different concentrations of the drug (n = 10). B Bacterial content in G. mellonella (n = 6)
3 Conclusion
Six previously unreported compounds, triterpenoids (1−4) and sterols (5 and 6) And 27 known compounds (7−33) were isolated and identified from C. coggygria. Anti-inflammatory bioactivity evaluation showed that compounds 5, 13, 15, 21, and 26 significantly inhibited NO production induced by LPS in RAW264.7 cells, with IC50 values ranging from 6.81 ± 0.15 And 21.54 ± 0.35 μM. The molecular docking results indicated the potential of NLRP3 and iNOS as target proteins for compound 15. Furthermore, the isolated triterpenoids and sterols were evaluated for their antibacterial activity against ten bacterial strains, revealing that compound 3 was the most active, with significant inhibitory activity against MRSA ATCC BAA-1717 (USA300). Subsequent analysis of the antibacterial mechanism indicated that compound 3 could potentially lead to the destruction of bacterial cell walls and membranes. Additionally, a synergistic effect was observed when 3 was combined with ampicillin. Compound 3 also exhibited antibacterial activity in a G. mellonella infection model. These findings provide a preliminary molecular and biological basis for the anti-inflammatory and antibacterial activities of small-molecule components in C. coggygria. In addition to being an ornamental tree species, C. coggygria may be developed as a value-added medicinal plant because of its highlighted antibacterial potential.
4 Experimental
4.1 General experimental procedures
The general experimental procedures were carried out in accordance with a previous report and are detailed in Text S1 [40].
4.2 Plant material
The tree stems and bark of C. coggygria were collected in October 2023 from the Tsinling Mountains of China. The plant species were identified by Dr. Zhen-Hai Wu, College of Life Sciences, Northwest A&F University. A voucher specimen (no. WUK 0480712) was deposited in the Herbarium of the College of Life Sciences, Northwest A&F University.
4.3 Extraction and isolation
The dried and crushed tree stems and bark of C. coggygria (30.0 kg) were subjected to three rounds of extraction with eight times their volume of heated 95% ethanol, resulting in a 4.5 kg crude extract. Subsequently, the crude extract was dissolved in three times the volume of water and partitioned sequentially with equal volumes of petroleum ether, and EtOAc, resulting in three partitions. Silica gel flash column chromatography (FCC) with dichloromethane/MeOH (80:1 to 2:1) was used to purify EtOAc fraction C (600.0 g), resulting in four fractions (A−D). Fraction A (30.0 g) was further purified using an FCC silica gel column and eluted with PE/EtOAc (40:1 to 0:1), resulting in five subfractions (A1−A5). Hereafter, A2 (1.4 g) was divided into four subfractions (A2-1−A2-4) using FCC silica gel with petroleum ether/EtOAc (20:1 to 2:1). The A2-1 (203.0 mg) and A2-2 (120.0 mg) fractions were separated by semipreparative HPLC to obtain compounds 3 (MeOH/H2O, 95:5; flow rate: 2 mL/min; 3.5 mg; tR = 22.2 min), 7 (MeOH/H2O, 95:5; flow rate: 2 mL/min; 8.8 mg; tR = 29.1 min), 16 (MeOH/H2O, 98:2; flow rate: 2 mL/min; 7.6 mg; tR = 18.2 min), and 17 (MeOH/H2O, 98:2; flow rate: 2 mL/min; 18.0 mg; tR = 25.0 min), respectively. Futher, Fraction A2-3 (60.0 g) was separated on a silica gel column eluted with PE/EtOAc (5:1) to give compounds 11 (10.5 mg) and 12 (7.6 mg). Fraction A3 (5.3 g) was separated on a silica gel column using PE/EtOAc (20:1 to 2:1) elution to yield subfractions A3-1 and A3-2. Fractions A3-1 (1.2 g) and A3-2 (1.9 g) were separated by chromatography on a Sephadex LH-20 column and eluted with dichloromethane/MeOH (1:1) and then purified using semi-preparative HPLC, which yielded compounds 4 (MeOH/H2O, 90:10; flow rate: 2 mL/min; 10.8 mg; tR = 20.1 min), 18 (MeOH/H2O, 90:10; flow rate: 2 mL/min; 12.0 mg; tR = 23.8 min), 19 (MeOH/H2O, 92:8; flow rate: 2 mL/min; 35.0 mg; tR = 23.1 min), 24 (MeOH/H2O, 92:8; flow rate: 2 mL/min; 26.0 mg; tR = 28.9 min), and 25 (MeOH/H2O, 92:8; flow rate: 2 mL/min; 8.0 mg; tR = 32.0 min). Fraction A4 (3.1 g) was separated on a silica gel column eluted with PE/EtOAc (20:1 to 1:1) to give fractions A4-1−A4-5. A4-1 (188.0 mg) was further separated on silica gel CC (PE/EtOAc, 12:1) followed by elution to yield 8 (18.4 mg), 9 (22.0 mg), and 15 (10.8 mg). Fraction A4-2 (550.0 mg), on the other hand, was subjected to chromatography on Sephadex LH-20 (Me2CO) and then purified using HPLC (MeOH/H2O, 89:11), which yielded compound 1 (3.3 mg, tR = 45.8 min) and 2 (2.8 mg, tR = 47.2 min). A4-3 (1.1 g) was applied to silica gel CC eluted with a gradient of PE/EtOAc (20:1 to 5:1), to give five subfractions (A4-3–1−A4-3–4). Section A4-3–2 (65.0 mg) was purified by semipreparative HPLC (MeOH/H2O, 85:15; flow rate: 2 mL/min) to yield compounds 5 (6.0 mg; tR = 26.6 min) and 6 (6.5 mg; tR = 28.0 min). Similarly, compounds 10 (4.5 mg; tR = 32.0 min) and 13 (9.8 mg; tR = 35.2 min) were isolated from subfraction A4-3–3 (320.0 mg) using semi-preparative HPLC (MeOH/H2O, 86:14; flow rate: 2 mL/min). As well, A4-3–4 (980.0 mg) was subjected to Sephadex LH-20 column elution with MeOH to obtain subfractions A4-3-4-1 and A4-3-4-2. Compound 14 (26.0 mg; tR = 24.3 min) was isolated from subfraction A4-3-4-1 (310.0 mg) by semipreparative HPLC (MeOH/H2O, 81:19; flow rate: 2 mL/min). Additionally, compounds 20 (4.5 mg; tR = 20.8 min), 21 (5.0 mg; tR = 34.5 min), and 26 (85.0 mg; tR = 38.0 min) were isolated from A4-3-4-2 (525.0 mg) by semipreparative HPLC (MeOH/H2O, 84:16; flow rate: 2 mL/min). Fraction B (15.0 g) was isolated using silica gel CC (PE/EtOAc, 20:1 to 1:1) to obtain three fractions (B1−B3). B1 (1.5 g) was then purified over a Sephadex LH-20 column and eluted with dichloromethane/MeOH to afford B1-1− B1-4. Separation of B1-2 (160.0 mg) on semipreparative HPLC (MeOH/H2O, 88:12; flow rate: 2 mL/min) afforded compounds 22 (12.8 mg; tR = 20.6 min) and 23 (3.8 mg; tR = 23.9 min). Fraction C (300.0 g) was separated on a silica gel column chromatograph using gradient elution (dichloromethane/MeOH, 40:1 to 5:1), yielding five subfractions (C1−C5). Fraction C2 (16.0 g) was further separated on a silica gel column and eluted with dichloromethane/MeOH (40:1 to 10:1) to obtain fractions C2-1−C2-6. Purification of fraction C2-2 (1.3 g) via semipreparative HPLC (MeOH/H2O, 74:26; flow rate: 2 mL/min) yielded compounds 27 (7.5 mg; tR = 19.2 min), 28 (4.6 mg; tR = 25.0 min), 31 (75.0 mg; tR = 27.6 min), 32 (86.0 mg; tR = 29.4 min), and 33 (24.5 mg; tR = 35.2 min). Fraction C2-3 (2.1 g) was purified by semi-preparative HPLC (MeOH/H2O, 76:24; flow rate: 2 mL/min) to yield compound 29 (19.0 mg, tR = 20.0 min)and 30 (46.0 mg; tR = 26.9 min).
Coggygrnoid A (1): Colorless solid;
Coggygrnoid B (2): Colorless solid;
Coggygrnoid C (3): Faint yellowish gum;
Coggygrnoid D (4): Yellowish gum;
Coggygrerol A (5): Yellowish gum;
Coggygrerol B (6): Yellowish gum;
4.4 Anti-inflammatory assay
As outlined in our previous article, the MTT assay and Griess reaction were employed to measure cell viability and NO production [41]. Futhermore, Molecular docking studies was operated as described previously [41].
4.5 Anti-bacterial assay
The effects of compounds 1−27 against the five gram-positive bacteria (Methicillin-resistant S. aureus 171, Methicillin-resistant S. aureus ATCC BAA-1717 (USA300), Methicillin-resistant S. aureus ATCC 12228, Methicillin-resistant S. aureus 209, and S. aureus ATCC 29213) and five gram-negative bacteria (E. coli CMCC 44103, E. coli CMCC 25922, Multi-drug-resistant P. aeruginosa 110, Multi-drug-resistant P. aeruginosa 174, and A. baumannii ATCC 19606) were evaluated in vitro. The minimal inhibitory concentration (MIC) was determined for compounds 1–27 using the antibacterial activity test protocols described in previous reports [42]. Based on previously reported methods [42], the growth curve of MRSA171 treated with compound 3 were evaluated. The previously described method was used to determine the time-kill curve of the test bacteria [43].
4.6 Determination of alkaline phosphatase (AKP) activity
Following an 8-h co-incubation of MRSA ATCC BAA-1717 (USA300) with 3, culture supernatants from each treatment group were collected. The concentration of alkaline phosphatase (AKP) was subsequently determined using a commercial AKP assay kit, following the manufacturer's protocol [44].
4.7 Assay for biofilm suppression
MRSA ATCC BAA-1717 (USA300) was incubated with 3 for 24 h. After incubation, the medium was removed And the biofilms were washed twice with PBS. Then, 0.1% crystal violet solution was added to each well And incubated for 30 min. The dye was dissolved by adding 33% glacial acetic acid after staining. After incubating for 1 h, the absorbance at OD590 was determined using a microplate reader [45].
4.8 Scanning electron microscopy (SEM)
According to our method, with minor modifications [42], a scanning electron microscopy (SEM) study was conducted to observe the cellular morphological changes in MRSA ATCC BAA-1717 (USA300) treated with 3. MRSA ATCC BAA-1717 (USA300) (approximately 2 × 108 CFU/mL) was treated with compound 3 at 4 × MIC. After culturing for 10 h at 37 ℃, MRSA ATCC BAA-1717 (USA300) was collected, washed with PBS (0.1 M, pH7.0) and then anchored with glutaraldehyde. Field-emission scanning electron microscopy (FE-SEM; Nova Nano SEM-450, FEI Instruments, Inc. Hillsboro, Oregon) was used for the microstructural evaluation of MRSA ATCC BAA-1717 (USA300).
4.9 Checkerboard MIC assay
The microdilution checkerboard method was used to evaluate the antibacterial effect of compound 3 in combination with ampicillin against MRSA ATCC BAA-1717 (USA300). Specifically, two-fold serial dilutions of 3 And ampicillin were prepared in a 96-well microtiter plate And inoculated with a bacterial suspension at a final concentration of 1× 108 CFU/mL. After incubation at 37 ℃ for 24 h, the interaction between the two agents was assessed using the fractional inhibitory concentration index (FICI) formula [46].

|
4.10 G. mellonella infection model and in vivo antibiotic activity assessment
G. mellonella larvae are used widely as surrogate infectious diseasemodels, due to ease ofuse and the presence of an innate immune system functionally similar to that of vertebrates [47]. To Analyze the survival of infected larvae, 5 μL of the bacterial inoculum (1 × 108 CFU/mL) was injected dorsolaterally into the hemocoel of the last-instar larvae. Following An incubation period at 37 ℃ for 60 min And observation that the larvae continued to survive normally, 5 μL of each working solution of compound 3 or the positive drugs (5, 10, And 20 mg/kg) were injected. The larvae were deemed deceased when they exhibited An absence of tactile response following a 72-h incubation at 37 ℃. Similarly, the second batch of G. mellonella was inoculated with bacteria And treated with different concentrations of the drug using the same method as above. After 24 h, the entire G. mellonella homogenate was suspended in PBS, then serially diluted tenfold to 108-fold. A 100 μL aliquot was spread onto agar plates, And colony counts were performed after 18 h (expressed in log10 CFU/g).
4.11 Statistical analysis
All of the biological experimental data presented in this paper were replicated three times, and the results are shown as the mean ± standard deviation (SD).
Notes
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (No. 32201249, 22277099, 22077102, 32070388), the Qin ChuangYuan "Scientists+Engineers" Team Construction Project (No. 2023KXJ-214), the Shaanxi Province Innovation Support Project (2025ZY1-GNYZ-13), the Shaanxi Administration of Traditional Chinese Medicine (No. 2022-09-20), and the Northwest A&F University "Double First-Class" Construction Project. The Advance Analysis and Measurement Center of Northwest A&F University helped us measure the NMR and MS spectra.
Author contributions
YT-Z: performing separation, purification and structural identification and wrote this manuscript. ZR-L: testing NMR and HRESIMS data. JH-L: testing the anti-inflammatory activity. RH-C and W-W: testing the anti-bacterial activit. YQ-G, CH-L and JM-G: designed this research work and provided funding support. All authors have read and agreed to publish the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (No. 32201249, Chun-Huan Li, 22277099, Jin-Ming Gao, 22077102, Jin-Ming Gao, 32070388, Jin-Ming Gao)
Data availability
Upon reasonable request, data utilised or analysed during this study may be obtained from the corresponding author.
Declarations
Competing interests
All the authors declare no competing financial interest.
References
-
1.Editorial Committee of the Flora of China, Chinese Academy of Sciences. Flora of China 1980;45: 96-99. PubMed Google Scholar
-
2.Tzakou I, Bazos A. Essential oils of leaves, inflorescences and infructescences of spontaneous Cotinus coggygria Scop. from Greece. Flavour Fragr J. 2005;20(5): 531-3. CrossRef PubMed Google Scholar
-
3.Antal DS, Schwaiger S, Ellmerer-Müller EP, Stuppner H. Cotinus coggygria wood: novel flavanone dimer and development of an HPLC/UV/MS method for the simultaneous determination of fourteen phenolic constituents. Planta Med 2010;76(15): 1765-72. CrossRef PubMed Google Scholar
-
4.Westenburg HE, Lee KJ, Lee SK, Fong HH, vanBreemen RB, Pezzuto JM, et al. Activity-guided isolation of antioxidative constituents of Cotinus coggygria. J Nat Prod 2000;63(12): 1696-8. CrossRef PubMed Google Scholar
-
5.Demirci BETÜL, Demirci FATİH, Başer KHC. Composition of the essential oil of Cotinus coggygria Scop. from Turkey. Flavour Fragr J. 2003;18(1): 43-4. CrossRef PubMed Google Scholar
-
6.Nićiforović N, Mihailović V, Mašković P, Solujić S, Stojković A, Muratspahić DP. Antioxidant activity of selected plant species; potential new sources of natural antioxidants. Food Chem Toxicol 2010;48(11): 3125-30. CrossRef PubMed Google Scholar
-
7.Ivanova D, Gerova D, Chervenkov T, Yankova T. Polyphenols and antioxidant capacity of Bulgarian medicinal plants. J Ethnopharmacol 2005;96(1–2): 145-50. CrossRef PubMed Google Scholar
-
8.Long L, Ma Z, Cui E, Han C, Zhang L, Zhang B. Research on depressurization effect of cotinus coggygria. Chin J Lab Diagn 2009;13: 330-1. PubMed Google Scholar
-
9.Constantin RP, Constantin J, Pagadigorria CLS, Ishii-Iwamoto EL, Bracht A, Ono MDKC, et al. The actions of fisetin on glucose metabolism in the rat liver. Cell Biochem Funct 2010;28(2): 149-58. CrossRef PubMed Google Scholar
-
10.Marčetić M, Božić D, Milenković M, Malešević N, Radulović S, Kovačević N. Antimicrobial, antioxidant and anti-inflammatory activity of young shoots of the smoke tree, Cotinus coggygria Scop. Phytother Res 2013;27(11): 1658-63. CrossRef PubMed Google Scholar
-
11.Liu WY, Qiu H, Li HM, Zhang R, Pan YK, Cao CY, et al. Prenylated flavonoids from Hypericum perforatum L. and their anti-neuroinflammatory and neuroprotective activities. Ind Crops Prod 2024;216: 118792. CrossRef PubMed Google Scholar
-
12.Liu WY, Gao LL, Zhou W, Ma YN, Tian JM, Gao JM. Hypertums a-j, bioactive polycyclic polyprenylated acylphloroglucinols with diverse skeletons from Hypericum perforatum L. (St. John's wort). Phytochemistry 2025;235: 114450. CrossRef PubMed Google Scholar
-
13.Liu WY, Lei XX, Wang WJ, Tian JM, Gao YQ, Gao JM. Hyperforatone A, the 1, 8-seco rearranged polycyclic polyprenylated acylphloroglucinol with a unique bicyclo [5.4.0] undecane core from Hypericum perforatum. Chin Chem Lett 2025;36(4): 110478. CrossRef PubMed Google Scholar
-
14.Tang JJ, Huang LF, Deng JL, Wang YM, Guo C, Peng XN, et al. Cognitive enhancement and neuroprotective effects of OABL, a sesquiterpene lactone in 5xFAD Alzheimer's disease mice model. Redox Biol 2022;50: 102229. CrossRef PubMed Google Scholar
-
15.Xie JY, Li P, Yan XT, Gao JM. Discovery from hypericum elatoides and synthesis of hyperelanitriles as α-aminopropionitrile-containing polycyclic polyprenylated acylphloroglucinols. Commun Chem 2024;7(1): 1-1. CrossRef PubMed Google Scholar
-
16.Xie JY, Gao YQ, Liu WY, Zhang SY, Li D, Wang WJ, Tang JJ, Gao JM. Hypertaxoids A and B, two unique meroterpenoids from Hypericum elatoides as microtubule stabilizers and their antiproliferative effects on cervical cancer. Chin Chem Lett 2025, 111580. CrossRef PubMed Google Scholar
-
17.Xie JY, Wang ZX, Liu WY, Liu HW, Li D, Sang YF, et al. Hyperelatolides A-D, antineuroinflammatory constituents with unusual carbon skeletons from Hypericum elatoides. J Nat Prod 2023;86(8): 1910-8. CrossRef PubMed Google Scholar
-
18.Quang DN, Long LD, Tung NQ, Thanh NN, Tham LX. Endertiins A-B, two lanostane triterpenoids from the fruit bodies of the mushoom Humphreya endertii. Nat Prod Res 2022;36(6): 748-53. CrossRef PubMed Google Scholar
-
19.Zhang F, Wang JS, Gu YC, Kong LY. Triterpenoids from Aglaia abbreviata and their cytotoxic activities. J Nat Prod 2010;73(12): 2042-6. CrossRef PubMed Google Scholar
-
20.Xu JL, Gu LH, Wang ZT, Bligh A, Han ZZ, Liu SJ. Seventeen steroids from the pith of Tetrapanax papyriferus. J Asian Nat Prod Res 2016;18(12): 1131-7. CrossRef PubMed Google Scholar
-
21.Makino M, Motegi T, Fujimoto Y. Tirucallane-type triterpenes from Juliania adstringens. Phytochemistry 2004;65(7): 891-6. CrossRef PubMed Google Scholar
-
22.Ono M, Yamashita M, Mori K, Masuoka C, Eto M, Kinjo J, et al. Sesquiterpenoids, triterpenoids, and flavonoids from the fruits of Schinus molle. Food Sci Technol Res 2008;14(5): 499-499. CrossRef PubMed Google Scholar
-
23.Mulholland DA, Nair JJ. Triterpenoids from Dysoxylum pettigrewianum. Phytochemistry 1994;37(5): 1409-11. CrossRef PubMed Google Scholar
-
24.Liu W, Gao J, Li M, Aisa HA, Yuan T. Tirucallane triterpenoids from the mastic (Pistacia lentiscus) and their anti-inflammatory and cytotoxic activities. Phytochemistry 2021;182: 112596. CrossRef PubMed Google Scholar
-
25.An XR, Feng YP, Sun YW, Yang X, Liu W, Yuan T. A new nortriterpenoid from mastic. China J Chin Mater Med 2024;49(23): 6342-51. PubMed Google Scholar
-
26.Tanaka R, Matsunaga S. Triterpene dienols and other constituents from the bark of Phyllanthus flexuosus. Phytochemistry 1988;27(7): 2273-7. CrossRef PubMed Google Scholar
-
27.Kajikawa M, Yamato KT, Fukuzawa H, Sakai Y, Uchida H, Ohyama K. Cloning and characterization of a cDNA encoding β-amyrin synthase from petroleum plant Euphorbia tirucalli L.. Phytochemistry 2005;66(15): 1759-66. CrossRef PubMed Google Scholar
-
28.Wen L, Weiming C, Zhi X, Xaotain L. New triterpenoids of Pholidota chinensis. Planta Med 1986;52(1): 4-6. CrossRef PubMed Google Scholar
-
29.Ferreira MJ, Lobo AM, Nascimento JM, Wyler H. Tetra-and pentacyclic triterpenes from the aerial parts of Euphorbia piscatoria. Planta Med 1994;60(6): 581-2. CrossRef PubMed Google Scholar
-
30.Shirane N, Murabayashi A, Masuko M, Uomori A, Yoshimura Y, Seo S, et al. Effect on ergosterol biosynthesis of a fungicide, SSF-109, in Botrytis cinerea. Phytochemistry 1990;29(8): 2513-20. CrossRef PubMed Google Scholar
-
31.Kim JS, Yean MH, Lee SY, Lee JH, Kang SS. Phytochemical studies on Lonicera caulis (3)-iridoids. Korean J Pharmacogn 2009;40(4): 334-8. PubMed Google Scholar
-
32.Li YH, Luo F, Peng SL, Liang J, Ding LS. A new dihydroisocoumarin from the rhizomes of Notopterygium forbesii. Nat Prod Res 2006;20(9): 860-5. CrossRef PubMed Google Scholar
-
33.Sun XB, Zhao PH, Xu YJ, Sun LM, Cao MA, Yuan CS. Chemical constituents from the roots of Polygonum bistorta. Chem Nat Compd 2007;43(5): 563-6. CrossRef PubMed Google Scholar
-
34.Gan KH, Kuo SH, Lin CN. Steroidal constituents of Ganoderma applanatum and Ganoderma neo-japonicum. J Nat Prod 1998;61(11): 1421-2. CrossRef PubMed Google Scholar
-
35.Prachayasittikul S, Buraparuangsang P, Worachartcheewan A, Isarankura-Na-Ayudhya C, Ruchirawat S, Prachayasittikul V. Antimicrobial and antioxidative activities of bioactive constituents from Hydnophytum formicarum Jack. Molecules 2008;13(4): 904-21. CrossRef PubMed Google Scholar
-
36.Junior GMV, deMSousa CM, Cavalheiro AJ, Lago JHG, Chaves MH. Phenolic derivatives from fruits of Dipteryx lacunifera Ducke and evaluation of their antiradical activities. Helv Chim Acta 2008;91(11): 2159-67. CrossRef PubMed Google Scholar
-
37.Zhou L, Li D, Wang J, Liu Y, Wu J. Antibacterial phenolic compounds from the spines of Gleditsia sinensis Lam. Nat Prod Res 2007;21(4): 283-91. CrossRef PubMed Google Scholar
-
38.Shrestha S, Lee DY, Park JH, Cho JG, Lee DS, Li B, et al. Flavonoids from the fruits of Nepalese sumac (Rhus parviflora) attenuate glutamate-induced neurotoxicity in HT22 cells. Food Sci Biotechnol 2013;22(4): 895-902. CrossRef PubMed Google Scholar
-
39.Haraguchi H, Tanaka Y, Kabbash A, Fujioka T, Ishizu T, Yagi A. Monoamine oxidase inhibitors from Gentiana lutea. Phytochemistry 2004;65(15): 2255-60. CrossRef PubMed Google Scholar
-
40.Shi YF, Zhu YT, Zhang ZH, Chen MS, Gao S, Zhang Q, et al. Structurally diverse chromane meroterpenoids from Rhododendron capitatum with multifunctional neuroprotective effects. Eur J Med Chem 2025;283: 117188. CrossRef PubMed Google Scholar
-
41.Yang JR, Zhu YT, Zeng YQ, Li HQ, Li CH, Gao JM. Three new ionone glycosides from rhododendron capitatum maxim. Molecules 2024;29(11): 2462. CrossRef PubMed Google Scholar
-
42.Chen XM, Lu W, Zhang ZH, Wang PP, Zhang XJ, Xiao CJ, et al. Discovery of diversified cassane diterpenoids as potent antibacterial agents from Caesaplinia pulcherrima and their mechanisms. Pest Manag Sci 2023;79(7): 2539-55. CrossRef PubMed Google Scholar
-
43.Guo W, Yang Z, Wang K, Li W, Zhao Y, Yang Y, et al. Discovery of unique bis-substituted aromatic amide derivatives as novel highly potent antibiotics for combating methicillin-resistant Staphylococcus aureus (MRSA). J Med Chem 2024;67(3): 2129-51. CrossRef PubMed Google Scholar
-
44.Ma CY, He N, Zhao YY, Xia DD, Wei JF, Kang WY. Antimicrobial mechanism of hydroquinone. Appl Biochem Biotechnol 2019;189: 1291-303. CrossRef PubMed Google Scholar
-
45.Long GQ, Li XM, Wang DD, Bao TRG, Yang YC, Zheng YY, et al. Prenylated flavonoids and derivatives isolated from the root barks of Sophora flavescens as potent antibacterial agents against Staphylococcus aureus. Ind Crop Prod 2022;189: 115834. CrossRef PubMed Google Scholar
-
46.Liu ZY, Zhou QY, Xue JJ, Cui MH, Xu L, Fang TQ, et al. Recent discoveries of propyl gallate restore the antibacterial effect of tigecycline against tet (X4)-positive Escherichia coli. Biochem Pharmacol 2025;231: 116638. CrossRef PubMed Google Scholar
-
47.Chang S, Li Y, Huang X, He N, Wang M, Wang J, et al. Bioactivity-based molecular networking-guided isolation of epicolidines A-C from the endophytic fungus Epicoccum sp. 1–042. J Nat Prod 2024;87(6): 1582-90. CrossRef PubMed Google Scholar
Copyright information
© The Author(s) 2026, modified publication 2026.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
