
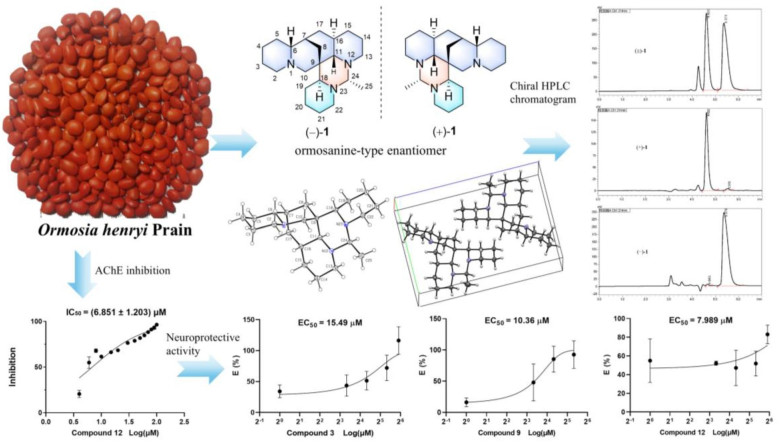
(+)-/(−)-Ormohenins A and B, two pairs of ormosanine-type enantiomers and their derivatives with neuroprotective activity from Ormosia henryi Prain

Abstract
Two pairs of undescribed alkaloid enantiomers, (+)-/(−)-ormohenins A (1) and B (2), were isolated from the seeds of Ormosia henryi Prain, along with four undescribed alkaloids (3, 4, 7 and 8) and seven known ones (5, 6, 9–13). Compounds 1–6 belong to the ormosanine-type alkaloids, compounds 7, 9, and 11 are of the lupinine-type, compounds 8 and 10 are classified as anagyrine-type alkaloids, 12 and 13 are cytisine-type alkaloids. The chemical structures of 1–13 were elucidated through comprehensive NMR and MS data analyses. Furthermore, the racemates (±)-1 and (±)-2 were successfully resolved into their respective optically pure enantiomers using a chiral HPLC system. The absolute configurations of compounds 1–3 were determined using single-crystal X-ray diffraction and corroborated by DFT calculations of specific rotations. The absolute configurations of 4, 7, and 8 were assigned by the experimental electronic circular dichroism (ECD) with those predicted using TDDFT calculations. Compound 12 exhibited significant acetylcholinesterase (AChE) inhibitory activity with the IC50 value of 6.581 ± 1.203 μM. The neuroprotective effects of these compounds against Aβ25-35 induced cell damage in PC12 cells were investigated, and compounds 3, 9, and 12 exhibited significant neuroprotective effects against Aβ25-35 induced PC12 cell damage, with the EC50 values of 7.99–15.49 μM, respectively.Graphical Abstract
Keywords
Ormosia henryi Ormosanine-type alkaloids Specific rotation calculations AChE inhibitory Neuroprotective activity1 Introduction
Alzheimer's disease (AD), the most common neurodegenerative disorder, is typified by the extracellular buildup of β-amyloid (Aβ) plaques and the intracellular formation of neurofibrillary tangles (NFTs), which are composed of hyperphosphorylated tau protein. Additionally, AD is marked by synaptic dysfunction, neuronal loss, and the activation of glial cells [1]. Current pharmacological interventions for Alzheimer's disease (AD) predominantly focus on alleviating symptoms. The arsenal includes acetylcholinesterase (AChE) inhibitors such as donepezil, rivastigmine, and galantamine, which aim to boost acetylcholine levels and thereby enhance cognitive function. Additionally, the N-methyl-D-aspartate (NMDA) receptor antagonist memantine is employed to modulate glutamate activity and protect neurons from overstimulation. More recently, anti-Aβ monoclonal antibodies like lecanemab and aducanumab have been introduced to target and clear Aβ plaques [2, 3]. While these medications can enhance cognitive abilities and daily functioning, or decelerate the pace of functional and cognitive deterioration, they are not capable of arresting the disease's progression. Given these limitations, there is an urgent need to identify and develop innovative therapeutic agents that can effectively address the underlying pathophysiology of AD and potentially alter its course.
Natural products, with their rich chemical diversity and unique structural features, have garnered significant attention for their potential to prevent neurodegeneration and enhance cognitive functions. Natural compounds often possess a wide array of pharmacological actions that can modulate multiple pathways implicated in neurodegenerative diseases. Their inherent complexity and multifunctionality enable them to interact with various biological targets, offering a holistic approach to combating neurodegeneration [4]. Ormosia henryi Prain, a member of the Ormosia genus, is a perennial evergreen tree commonly found in the southern regions of China [5]. This plant has a long history of use in traditional Chinese folk medicine, where its roots, leaves and stem bark have been applied to treat swallowing disorders, pain, and inflammation [6]. Clinical observations suggest that the leaves of O. henryi Prain have the effects of refreshing, invigorating, and antidepressant, hinting at its potential in the treatment of depression [7, 8]. Despite these promising traditional applications, there has been a dearth of scientific research into the plant's chemical constituents and pharmacological properties. Preliminary studies have identified the presence of flavonoids [9] and alkaloids [10, 11] in O. henryi Prain. As a potential renewable resource, a more in-depth investigation into the chemical composition and pharmacological activities of O. henryi Prain is warranted. In our recent research, we have successfully isolated two pairs of new alkaloid enantiomers, namely (+)-/(−)-ormohenins A (1) and B (2), along with four new alkaloids, (−)-ormohenin C (3), (−)-ormohenin D (4), (6R, 7S, 9S, 11R)-4, 5-dehydro-α-isolupanine (7), and (7S, 9S, 11S)-15, 16-dehydroanagyrine (8). We also identified seven known alkaloids, namely (±)-18-epiormosanine (5), (±)-ormosanine (6), (+)-lupanine (9), (+)-anagyrine (10), (+)-α-isolupanine (11), (+)-cytisine (12), and (+)-N-methylcytisine (13) (Fig. 1).
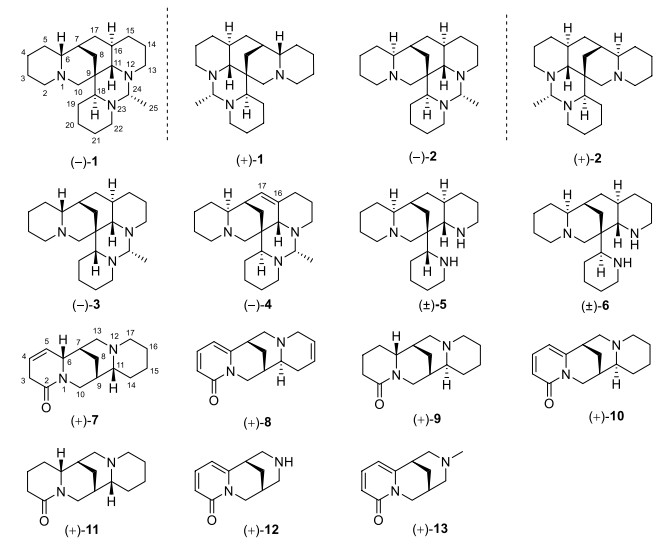
Chemical structures of 1–13
2 Results and discussion
Compound 1 was isolated as colorless crystals. Its molecular formula was determined to be C22H37N3, with six double bond equivalents (DBEs), based on the HRESIMS ion [M + H]+ at m/z 344.3061 (calcd for 344.3065). The 1H NMR and 13C NMR spectrum (Tables 1 and 3) revealed 22 carbon signals for compound 1, consisting of a doublet methyl group [δH-25 1.23 (d, J = 5.5 Hz), δC-25 17.0], 14 methylenes, six methines, and a nonprotonated carbon (δC-9 37.3). The 1H-1H COSY spectrum displayed three spin–spin systems: H-2/H-3/H-4/H-5/H-6/H-7/H-17(H-8)/H-16/H-15(H-11)/H-14/H-13, H-18/H-19/H-20/H-21/H-22, and H-24/H-25 (Fig. 2A). HMBC correlations (Fig. 2A) from H-6 to C-2, C-5, C-8 and C-10 confirmed that rings A and B were fused via N-1 and C-6. Further HMBCs from H-11 to C-8, C-10, C-13, C-16, and C-17 indicated rings B and D were connected through ring C. Ring E was positioned adjacent to rings C and D, as evidenced by HMBC correlations of H-11/C-8, C-10, C-13, C-24, and H-24/C-13. The connectivity of ring F to ring E was corroborated by the HMBC cross-peaks of H-18/C-9, C-24, and H-22/C-24. The planar structure of 1 was closely related to the known ormosia-type quinolizidine alkaloid, homoormosanine [10]. The primary difference in their 1D NMR spectra between them was the presence of an additional doublet methyl (C-25) signal in 1, which was assignable to C-24 supported by the HMBC correlations from CH3-25 to C-24.
1H NMR Spectroscopic Data (600 MHz) for 1–4 in CD3OD
1H NMR Spectroscopic Data (600 MHz) for 7 and 8 in CD3OD
13C NMR Spectroscopic Data (150 MHz) for 1–4, 7 and 8 in CD3OD
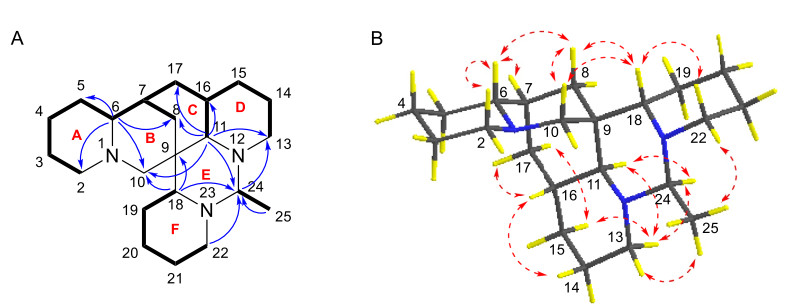
A 1H-1H COSYs (—) and selected HMBCs (→) of 1.B Key ROESY correlations (↔) of 1
The relative configuration of 1 was elucidated through analysis of its ROESY spectrum (Fig. 2B). Key cross-peaks of H-6/H-7, H-6/H-8β, H-8β/H-10β, H-15β/H-13β, H-13β/H-11, H-11/H-24, H-15β/H-17β suggested that these hydrogens, especially H-6, H-7, H-11, and H-24, were cofacial and arbitrarily assigned as β-orientation. The correlations of H-18/H-10β, H-18/H-8β, were observed, which suggested that these hydrogens were spatially nearby, especially inferred the α-orientation of H-18. The ROESY crosspeaks of H-18/H-22α, H-16/H-14α, H-16/H-17α, and H-13α/CH3-25 suggested that these hydrogens were α-orientated.
Compound 2 was obtained as colorless crystals. Its HRESIMS data exhibited the [M + H]+ ion peak at m/z 344.3062 (calcd for 344.3065), consistent with a molecular formula of C22H37N3. The molecular formula of compound 2, along with its hydrogen and carbon chemical shifts and the types of carbon atoms (Tables 1 and 3), are essentially the same as those of compound 1, which allows us to infer that the gross structure of compound 2 is similar to that of compound 1. The variations in some chemical shifts observed between them may be attributed to their differing stereoconfigurations.
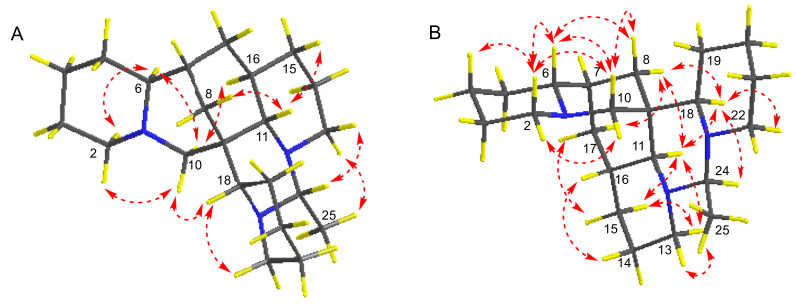
The analysis of the relative configuration of compound 2 was hindered due to the overlapping proton signals of H-6, H-11, H-16, and other critical hydrogens in its ROESY spectrum. This overlap can complicate the determination of the relative configuration based solely on ROESY data (Fig. 3A). However, the configuration for compounds 1 and 2 were successfully determined through single-crystal X-ray diffraction experiments. Consequently, the structures with relative configurations of 1 and 2 were unambiguously determined as depicted in Figs. 4A and 5B. Further analysis of the X-ray data revealed that both 1 and 2 possessed a centrosymmetric space group P-1, which is indicative of their racemic nature (Figs. 4A, 5B, and Tables S1, S2). The optical values of zero for compounds 1 and 2 also implied that they were racemic mixtures. Fortunately, chiral separations of compounds 1 and 2 were successfully performed using Chiralpak columns, resulting in the isolation of their optically pure enantiomers (Figs. 6 and 7).
Key ROESY correlations (↔) of 2 (A) and 3 (B)
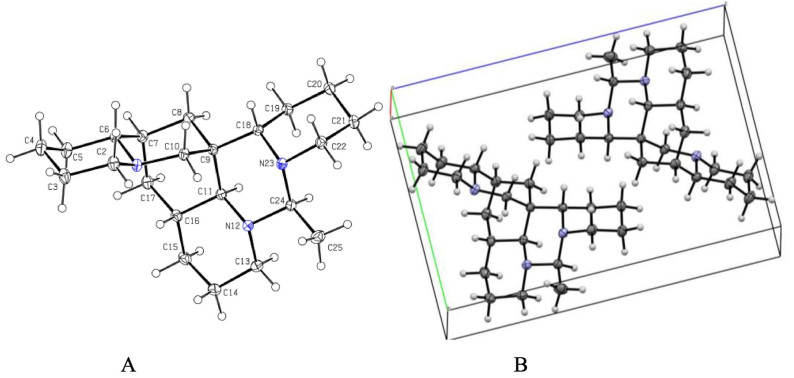
A X-ray crystal structure of 1.B Assembly of molecules of 1 in the crystals
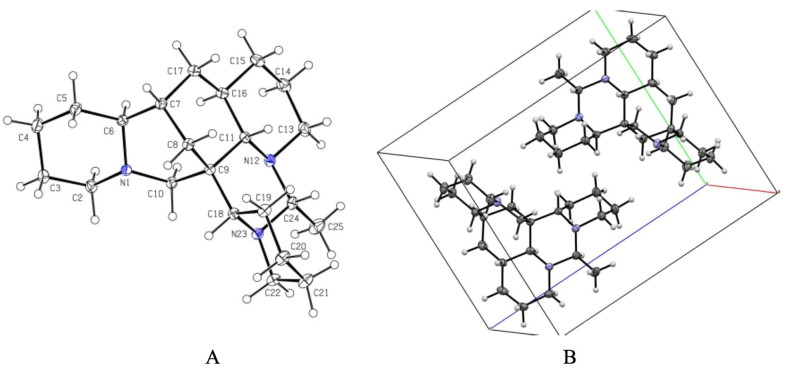
A X-ray crystal structure of 2.B Assembly of molecules of 2 in the crystals
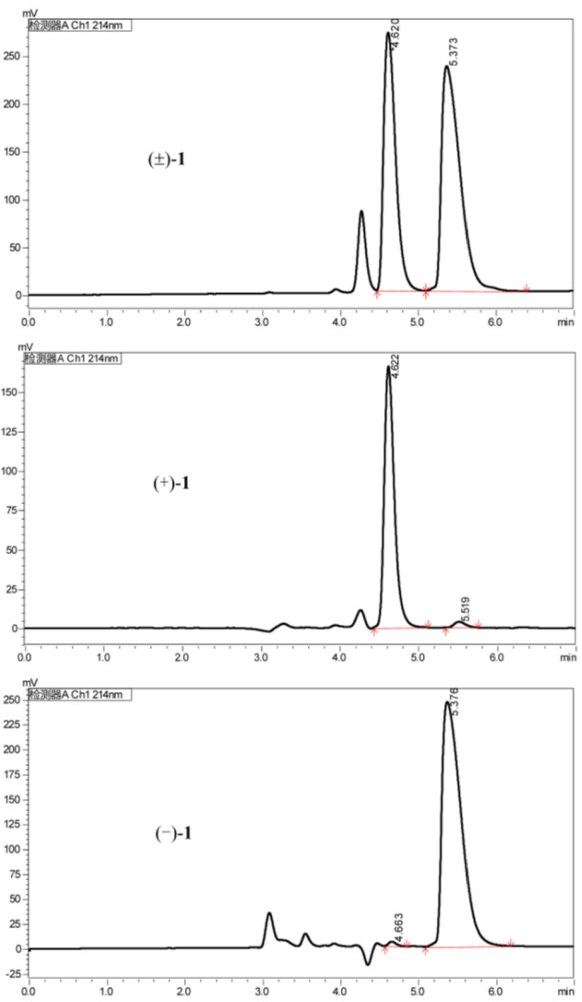
Chiral HPLC chromatogram of racemic 1 using a Chiralpak IE (IE00CE-SB022, 0.46 × 25 cm); isocratic elution with hexane/EtOH/DEA (80/20/0.1, v/v/v), flow rate 1 mL/min, UV detection at 214 nm
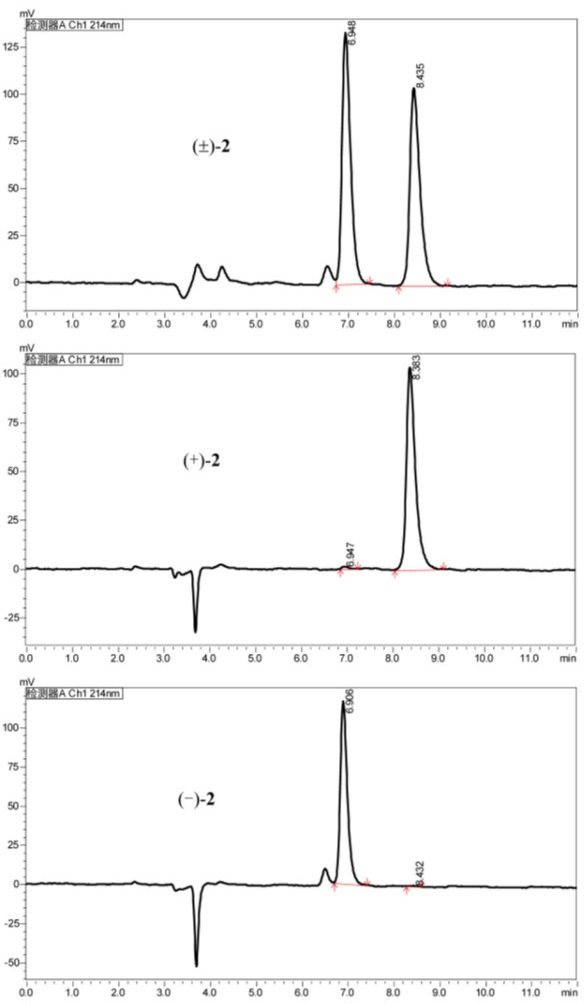
Chiral HPLC chromatogram of racemic 2 using a Chiralpak IF (IF00CE-RL017, 0.46 × 25 cm); isocratic elution with MeOH/DEA (100/0.1, v/v), flow rate 1 mL/min, UV detection at 214 nm
Although the separation of racemates 1 and 2 was successfully achieved, pinpointing their absolute configurations remained a challenge owing to their lack of qualified crystals and chromophores in the molecules. It is well-documented that the calculation of specific rotation is a valuable technique for elucidating the absolute structure of chiral compounds [12]. Consequently, we proceeded to determine the specific rotations of the resulting chiral HPLC fractions. For 1, chiral HPLC fraction Ⅰ at a retention time (tR) of 4.62 min exhibited a specific rotation of
Compound 3 was isolated as colorless crystals, and its molecular formula was determined to be C22H37N3 based on the HRESIMS data, which showed an [M + H]+ ion peak at m/z 344.3060 (calcd for 344.3065). The 1D NMR spectroscopic data (Tables 1 and 3) for 3 were consistent with those of 1, suggesting that 3 also shared the same planar structure as 1. The relative configuration of 3 was established as shown in Fig. 3B by ROESY correlations of H-2β/H-4, H-6/H-2β, H-6/H-10β, H-6/H-8β, H-10β/H-2β, H-10β/H-8β, H-8α/H-11, H-2α/H-10α, H-8α/H-18, H-8α/H-17β, H-18/H-11, H-18/H-24, H-18/H-22β, H-11/H-13β, H-13β/H-15β, H-11/H-15β, H-16/H-14α, H-17α/H-15α, CH3-25/H-13α. The absolute configuration of 3 was finally determined by single crystal X-ray diffraction analysis with CuKα radiation (Fig. 8). The Flack parameter of 0.04 (6) [13] established unambiguously the absolute configuration of 3 as 6R, 7R, 9S, 11S, 16R, 18R, 24R, and the compound was named ormohenin C.
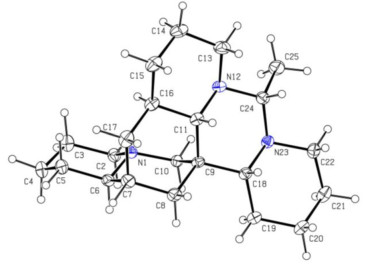
X-ray crystal structure of 3
Compound 4 was isolated as a yellowish oil. Its molecular formula was determined to be C22H35N3 with seven DBEs, based on the HRESIMS ion [M + H]+ at m/z 342.2898 (calcd for 342.2909). The NMR data (Tables 1 and 3) revealed 22 carbon signals, including four N-methylenes, four N-methines, a doublet methyl [δH-25 1.23 (d, J = 5.8 Hz), δC-25 16.1], and a nonprotonated carbon (δC-9 38.5). These features suggested that compound 4 is a structural congener of compounds 1–3. The primary distinction lies in the presence of an olefinic proton at δH-17 5.48, corresponding to a double bond (δC 129.0 and 134.5) in 4. The HMBC correlations (Fig. 9A) from H-17 to C-7, C-8, C-11, and C-15 assigned the Δ16, 17 double bond. The 1H-1H COSY spectrum (Fig. 9A) also confirmed the Δ16, 17 double bond and displayed three spin–spin systems: H-2/H-3/H-4/H-5/H-6/H-7/H-17(H-8)/H-16/H-15(H-11)/H-14/H-13, H-18/H-19/H-20/H-21/H-22, and H-24/H-25. The other HMBCs of H-7/C-5, H-6/C-5, H-6/C-10, H-4/C-6, H-2/C-6, H-10/C-2, H-10/C-6, H-10/C-9, H-10/C-8, H-10/C-11, H-13/C-11, H-13/C-14, H-18/C-11, H-18/C-19, H-22/C-18, H-24/C-11, H-24/C-18, H-24/C-13, and H-25/C-24 further determined the structure of 4 as shown. The relative configuration of 4 was established by key ROESY correlations (Fig. 9B) of H-8β/H-18α, H-8α/H-11β, H-6/H-2α, H-6/H-10α, H-7/CH2-5, H-10β/H-2β, H-10β/H-18, H-18/H-19α, H-18/H-20α, H-11/H-13β, H-19β/H-24, H-24/H-11β, H-24/H-13β, CH3-25/H-13α. The theoretical ECD spectrum of (6S, 7R, 9S, 11S, 18S, 24R)-4 was in good accordance with its experimental ECD spectrum (Fig. 10A), which allowed its absolute configuration as 6S, 7R, 9S, 11S, 18S, 24R. Consequently, compound 4 was designated as ormohenin D.
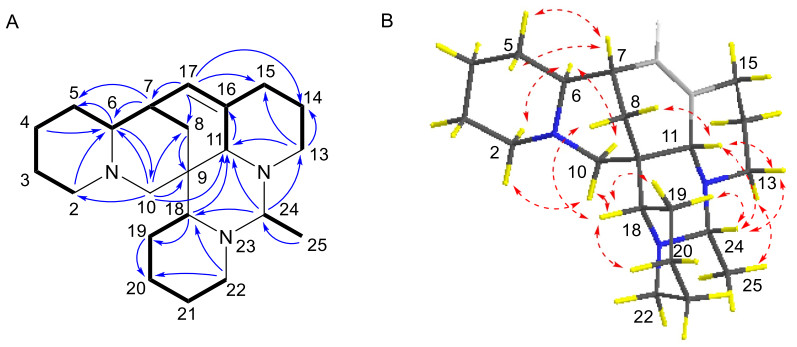
A 1H-1H COSYs (—) and selected HMBC correlations (→) of 4.B Key ROESY correlations (↔) of 4
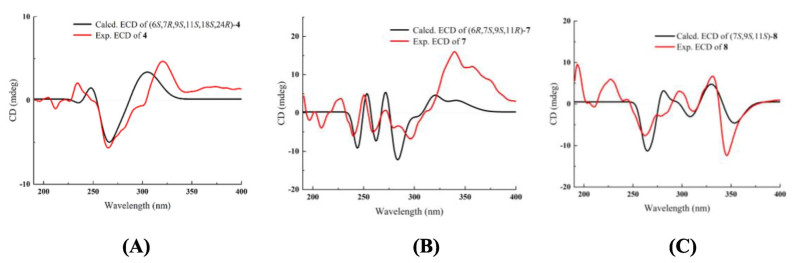
Experimental ECD and calculated ECD spectra of compounds 4, 7, and 8
Compound 7 was isolated as a yellowish oil, with a molecular formula of C15H22N2O, as determined by HRESIMS from a quasi-molecular ion [M + H]+ at m/z 247.1799 (calcd for 247.1820), corresponding to six DBEs. The 1H NMR and 13C NMR spectra (Tables 2 and 3) revealed the presence of 15 carbon signals, consisting of eight methylenes, six methines [including two olefinic methines (δH-4 5.82 br d, J = 10.2 Hz, δC-4 123.3; δH-5 5.54 br d, J = 10.2 Hz, δC-5 125.6)], and an amide carbonyl at δC-2 170.0. The presence of these functional groups, along with the remaining four DBEs, suggested that 7 was a tetracyclic alkaloid [14].
Compound 7 shared the same tetracyclic framework as α-isolupanine (11) [11], with the primary difference being the presence of olefinic signals in 7. The 1H-1H COSY spectrum confirmed the Δ4, 5 double bond and displayed a spin–spin system of H-3/H-4/H-5/H-6/H-7/H-8(H-13)/H-9/H-11(H-10)/H-14/H-15/H-16/H-17 (Fig. 11A). HMBC correlations from olefinic proton at δH-4 5.82 to C-2 (δC 170.0) and C-3 (δC 32.4), and from the olefinic proton at δH-5 5.54 to C-3 (δC 32.4) and C-6 (δC 63.0), also indicated that this double bond was located between C-4 and C-5 (Fig. 11A). The relative configuration of 7 was established through analysis of its ROESY spectrum (Fig. 11B). Key ROESY correlations of H-5/H-7, H-6/H-7, H-6/H-8β, H-6/H-10β, CH2-10/H-9 suggested that H-6, H-7, H-8β, H-9, and H-10β were cofacial and arbitrarily assigned β-orientation. Additionally, correlations of H-10α/H-14α, H-11/H-8α, H-11/H-14β, H-11/H-13β, H-11/H-17β were observed, suggesting that H-11 were β-oriented. The absolute configuration of 7 was determined by the experimental ECD and calculated ECD spectra. The calculated ECD spectrum of (6R, 7S, 9S, 11R)-7 matched very well with the experimental ECD spectrum (Fig. 10B). Therefore, compound 7 was identified as (6R, 7S, 9S, 11R)-4, 5-dehydro-α-isolupanine.
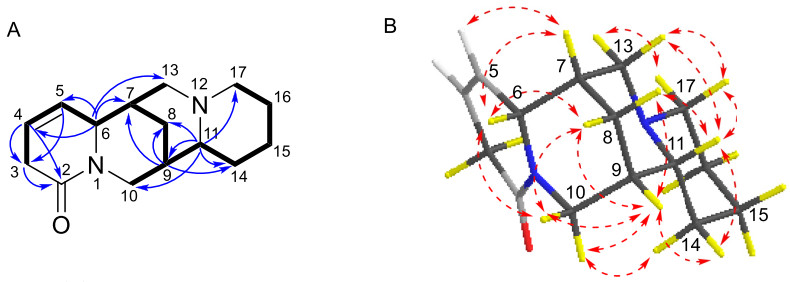
A 1H-1H COSY (—) and selected HMBC correlations (→) of 7.B Key ROESY correlations (↔) of 7
Compound 8 emerged as a new natural constituent, isolated as yellowish oil. Its molecular formula was deduced to be C15H18N2O, supported by HR-ESI-MS data exhibiting a quasi-molecular ion [M + H]+ at m/z 243.1491 (calcd 243.1492). The 1D and 2D NMR spectroscopic data of 8 (Tables 2 and 3), including ROESY data, were found to be identical to those of a synthetic intermediate S5 [15], suggesting that both compounds share the same planar structure and relative configurations. The specific rotation of 8 was determined to be
Seven known alkaloids were also isolated. Compounds 5 and 6 were obtained as crystals and identified as enantiomers, as evidenced by their specific rotations of zero. By comparing their spectroscopic data and optical rotations with the reported data in the literatures, the structures of known ones were unambiguously identified as (±)-18-epiormosanine (5) [16–18], (±)-ormosanine (6) [10, 18], (+)-lupanine (9) [10], (+)-anagyrine (10) [19–21], (+)-α-isolupanine (11) [11], (+)-cytisine (12) [22], and (+)-N-methylcytisine (13) [22].
The first and the main class of drugs currently used for AD are the AChE inhibitors and are widely used for the symptomatic treatment of mild-moderate AD [2]. All alkaloids were screened toward AChE via improved Ellman's method (Supporting Information). Compound 12 displayed significant AChE inhibitory activity, with an IC50 value of 6.851 ± 1.203 μM. Huperzine A was used as a positive control, with an IC50 value of 1.173 ± 0.223 μM.
Meanwhile, all isolated compounds were evaluated for their neuroprotective potency against the Aβ25-35 induced PC12 cell death by MTT assay. The result (Table 4 and Supporting Information) showed that compounds 3, 9, and 12 exhibited neuroprotective effects against Aβ25-35 induced PC12 cell damage. Among them, Compound 12 displayed the most neuroprotective activity, with an EC50 value of 7.99 μM. Resveratrol was used as a positive control [23], with an EC50 value of 5.99 μM.
The neuroprotective effects of compounds 1–12 against Aβ25-35-induced cell damage in PC12 cells
3 Experimental section
3.1 General experimental procedures
Optical rotations were measured using a Rudolph Research Analytical Autopol Ⅵ automatic polarimeter (Rudolph Research Analytical, NJ, USA). Melting points were determined with a YRT-3 melting point apparatus (Tianjin, China). UV spectra were recorded on an Evolution 300 PC UV–visible spectrophotometer (Thermo, USA). IR spectra were obtained using a Thermo Fisher IS50 spectrometer (Nicolet, USA) and a Bruker VERTEX 70 spectrometer (Germany), with samples prepared as KBr disks. ECD spectra were recorded on a Bio-Logic Science MOS-500 spectrometer. 1D and 2D NMR spectra were acquired on Bruker AV4600 NMR spectrometers, with TMS serving as the internal standard. HR-ESI–MS analyses were performed on Agilent 6545 and G6230A mass spectrometers. Single-crystal-X-ray diffractions were conducted using a Bruker APEX-Ⅱ CCD detector with CuKα and MoKα radiations. Semi-preparative HPLC was carried out on a Waters 1525 pump equipped with a Waters 2489 detector and a YMC-Pack ODS-A column (250 × 10 mm, S-5 μm, 12 nm, Japan). Chiral HPLC separations were performed on a Shimadzu LC-20AD system using CHIRALPAK IE (IE00CE-SB022, 0.46 × 25 cm) and CHIRALPAK IF (IF00CE-RL017, 0.46 × 25 cm) columns. Normal phase column chromatography was conducted using silica gel (100–200 mesh). Reversed-phase column chromatography employed C18 silica gel (150–200 mesh, Merck) and Sephadex LH-20 (Amersham Biosciences). Precoated silica gel GF254 plates (Qingdao Marine Chemical Plant, Qingdao, China) were utilized for TLC analysis.
3.2 Plant material
The seeds of O. henryi Prain were collected from Lishui of Zhejiang Province, China. A voucher sample (HLM-2020511s) was deposited in College of Xingzhi, Zhejiang Normal University.
3.3 Extraction and isolation
The air-dried powders of the seeds of O. henryi Prain (2.0 kg) were extracted three times with 95% EtOH at ambient temperature. The crude extract (144.1 g) was dissolved in water, adjusted to pH 2 with 2% H2SO4, and then adjusted to pH 11 with 2 mol/L NaOH. Subsequent extraction with a CHCl3/H2O soluble system provided a crude CHCl3 extract (87.6 g). The crude extract was subjected to silica gel column chromatography (CC) and eluted with a gradient of CHCl3/MeOH (50: 1–1: 1) to yield three fractions (Fr. A–C).
Fr. A (25.8 g) was further separated using C18 reversed-phase silica and eluted with a gradient of H2O/MeOH (20–90%), resulting in subfractions A1–A4. Fr. A1 (1.8 g) was separated using silica gel CC with a CHCl3/MeOH gradient (100: 1–1: 1) to yield A1a and A1b. Fr. A1a (0.8 g) was further separated by passage over an Sephadex LH-20 CC (EtOH) and silica gel CC (CHCl3/MeOH, 100: 1–10: 1) to give subfractions A1a2a and A1a2b. Compounds 7 (tR = 28 min, 39.8 mg), 8 (tR = 34 min, 6.5 mg), 9 (tR = 30 min, 78.8 mg) and 10 (tR = 52 min, 5.4 mg) were purified from fr. A1a2a by semipreparative HPLC with an eluent of 20% ACN/H2O. Fr. A2 (7.8 g) was processed through silica gel CC, C18 reversed-phase silica, and LH-20 CC to yield four subfractions, A2b1a–A2b1d. Compound 5 (8.1 mg) was obtained from the fr. A2b1a (0.4 g) by silica gel CC and crystallization in MeOH. Fr. A2b1c (0.5 g) was separated into two subfractions, A2b1c1A and A2b1c1B, using silica gel CC and C18 reversed-phase silica. 2 (78.1 mg) was obtained by crystallization of fr. A2b1c1B in MeOH. Fr. A4 (5.1 g) was separated using silica gel CC with a CHCl3/MeOH gradient (100: 1–10: 1) to yield four subfractions, A4a–A4d. Fr. A4a (0.9 g) was further separated over an Sephadex LH-20 CC and C18 reversed-phase CC to give three subfractions, A4a1-A4a3. Semi-preparative HPLC of fr. A4a3 (0.16 g) with 90% ACN/H2O yielded compounds 1 (tR = 55 min, 52.0 mg) and 3 (tR = 59 min, 42.9 mg). Semi-preparative HPLC of fr. A4a2 (0.28 g) with 87.5% MeOH/H2O yielded 4 (tR = 45.5 min, 46.0 mg). Compound 6 (26.1 mg) was obtained by crystallization of fr. A4b in MeOH. Compound 11 (38 mg) was obtained from fr. A4c by silica gel CC and Sephadex LH-20 CC, respectively.
Compound 12 (154 mg) was obtained from fr. C (18.5 g) by silica gel CC and Semi-preparative HPLC (50% MeOH/H2O, tR = 9.0 min). Fr. B (2.25 g) was further separated using C18 reversed-phase silica (H2O/MeOH 20–90%) and then silica gel CC (CH2Cl2/MeOH) to yield 13 (123 mg).
Subsequently separation of 1 by semi-preparative HPLC using a normal phase chiral column (Chiralpak IE) and eluting with n-hexane/EtOH/DEA (80: 20: 0.1) obtained (+)-1 (flow rate 1.0 mL/min, tR = 4.62 min) and (−)-1 (tR = 5.37 min), respectively. Separation of 2 by semi-preparative HPLC using a reverse phase chiral column (Chiralpak IF) and eluting with EtOH/DEA (100: 0.1) obtained (+)-2 (flow rate 1.0 mL/min, tR = 6.91 min) and (−)-2 (tR = 8.38 min), respectively.
3.4 Spectroscopic data of compounds
3.4.1 (±)-Ormohenin A (1)
Colorless needles (MeOH); m.p. 143.9 ℃;
(−)-1: colorless oil;
(+)-1: colorless oil;
3.4.2 (±)-Ormohenin B (2)
Colorless needles (MeOH); m.p. 145.1 ℃
(−)-2: colorless oil;
(+)-2: colorless oil;
3.4.3 Ormohenin C (3)
Colorless needles (MeOH); m.p. 140.0 ℃;
3.4.4 Ormohenin (4)
Yellowish oil;
3.4.5 (6R,7S,9S,11R)-4,5-Dehydro-α-isolupanine (7)
Yellowish oil;
3.4.6 (7S,9S,11S)-15,16-Dehydroanagyrine (8)
Yellowish oil;
3.4.7 The optical rotations of 5, 6, 9–13
18-Epiormosanine (5): colorless needles (MeOH); m.p. 220.0 ~ 221.0 ℃;
3.5 Crystallographic data
Compounds 1–3 were recrystallized from MeOH to give colorless needles at room temperature. Crystal data was obtained on a Bruker APEX-Ⅱ CCD diffractometer employing graphite monochromated Cu-Kα radiation (λ = 1.54178 Å) or Mo-Kα radiation (λ = 0.71073 Å) at 170.0 K. Using Olex2 [25], the structure was solved with the ShelXT [26] structure solution program using Intrinsic Phasing and refined with the ShelXL refinement package using Least Squares minimization. Crystallographic data (excluding structure factor tables) for 1–3 have been deposited at the Cambridge Crystallographic Data Center (CCDC number 2457561 for 1, deposition number: 2457563 for 2, 2, 457, 567 for 3. Copies of the data can be obtained free of charge via the internet at www.ccdc.cam.ac.uk/conts or upon application to CCDC, 12, Union Road, Cambridge CB21EZ, UK [tel: (+ 44) 1223-336-408; Fax: (+ 44) 1223-336-033; e-mail: deposit@ccdc.cam.ac.uk].
Ormohenin A (1): m.p. 143.9 ℃. Crystal data for C22H37N3 (M = 343.54 g/mol): triclinic, space group P-1 (no. 2), a = 6.8445(4) Å, b = 9.8999(5) Å, c = 14.9406(8) Å, α = 88.184(2)°, β = 83.162(2)°, γ = 69.941(2)°, V = 944.15(9) Å3, Z = 2, T = 170.0 K, μ(CuKα) = 0.534 mm−1, Dcalc = 1.208 g/cm3, 26, 269 reflections measured (5.958° ≤ 2Θ ≤ 137.378°), 3461 unique (Rint = 0.0299, Rsigma = 0.0223) which were used in all calculations. The final R1 was 0.0389 (I > 2σ(I)) and wR2 was 0.1026 (all data).
Ormohenin B (2): m.p. 145.1 ℃. Crystal data for C22H37N3 (M = 343.54 g/mol): triclinic, space group P-1 (no. 2), a = 6.8085(2) Å, b = 10.6845(4) Å, c = 13.8387(5) Å, α = 90.243(2)°, β = 98.8520(10)°, γ = 106.3930(10)°, V = 953.07(6) Å3, Z = 2, T = 170.0 K, μ(MoKα) = 0.070 mm−1, Dcalc = 1.197 g/cm3, 24, 182 reflections measured (4.852° ≤ 2Θ ≤ 67.15°), 6459 unique (Rint = 0.0308, Rsigma = 0.0337) which were used in all calculations. The final R1 was 0.0533 (I > 2σ(I)) and wR2 was 0.1453 (all data).
Ormohenin C (3): m.p. 143.9 ℃. Crystal data for C22H37N3 (M = 343.54 g/mol): orthorhombic, space group P212121 (no. 19), a = 9.3517(2) Å, b = 14.1492(3) Å, c = 14.8653(3) Å, V = 1966.96(7) Å3, Z = 4, T = 170.0 K, μ(CuKα) = 0.513 mm−1, Dcalc = 1.160 g/cm3, 20, 384 reflections measured (8.628° ≤ 2Θ ≤ 136.442°), 3585 unique (Rint = 0.0243, Rsigma = 0.0174) which were used in all calculations. The final R1 was 0.0317 (I > 2σ(I)) and wR2 was 0.0838 (all data). Flack parameter = 0.04(6).
3.6 Specific rotation and ECD calculations
Density functional theory (DFT) calculations of specific rotation and ECD are described in the Supporting Information.
3.7 AChE inhibitory activity assay
The AChE inhibitory activity of isolated compounds was assessed with a modified spectrophotometric method [27, 28], using huperzine A as the positive controls (see details in the Supporting Information).
3.8 Neuroprotective activity assay
A cell viability assay was conducted to test the cytotoxic effects of the compounds on PC12 cells. Meanwhile, the neuroprotective activity of all compounds against the Aβ25-35 induced PC12 cell death by MTT assay. PC12 cells were divided into four groups: normal control, model, positive control (Resveratrol), and isolates intervention groups (1, 10, 20, 40, 60, 80, 100 μmol/L). Detailed experimental procedures and experimental data can be found in the Supporting Information.
4 Conclusion
Ormosia plants are rich in alkaloids, which can be classified into ormosanine-type, lupinine-type, anagyrine-type, sparteine-type, cytisine-type, and cytisine-like-type [17, 24]. Investigation of the seeds of O. henryi Prain led to the isolation of 13 alkaloids. Compounds 1–4 are the first example of ormosanine-type alkaloids with a methylpyrimidine ring that are abundant in the seeds. They have no polar groups and have little polarity. Compounds 1, 2, 5 and 6 exist in the form of racemates. Pharmacological activity studies showed that cytisine (12) exhibited the most potent AChE inhibitory activity with an IC50 value of 6.851 ± 1.203 μM and the most excellent neuroprotective effects against Aβ25-35 induced PC12 cell damage, with an EC50 values of 7.99 μM. The new ormosanine-type alkaloid 3 exhibited moderate AChE inhibitory activity with an IC50 value of 141.5 ± 19.55 μM and significant neuroprotective effects against Aβ25-35 induced PC12 cell damage, with an EC50 values of 15.49 μM. This research enriched the structural diversity of alkaloids from the plants of the Ormosia genus and presented potent natural AChE inhibitors and neuroprotective compounds for further investigation.
Notes
Acknowledgements
This research was supported by grants from the National Natural Science Foundation of China (82003612) and the Startup Funding from Xingzhi College of Zhejiang Normal University (ZC302925002).
Author contributions
Ming Cheng: Methodology, Validation, Formal analysis, Investigation, Data curation, Wring—original draft. Xian-Si Zeng: Methodology, Supervision, Writing—review and editing. Zhao-Yun Yin: Validation, Formal analysis, Investigation, Data curation. Xiao-Yan Xie: Methodology, Formal analysis, Investigation, Data curation, Wring—original draft. Jia-Wen Zhu: Formal analysis, Investigation, Data curation. Jian-Feng Wang: Investigation. Ying-Kun Sheng: Investigation. Jin-Biao Xu: Conceptualization, Methodology, Supervision, Project administration, Resources, Data curation, Writing—review and editing.
Data availability
All data generated and analyzed during this study are included in this published article and its Additional file 1.
Declarations
Competing interests
These authors declare no conflict of interest in this study.
References
-
1.Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179(2): 312-39. CrossRef PubMed Google Scholar
-
2.Kim AY, Al Jerdi S, MacDonald R, Triggle CR. Alzheimer's disease and its treatment-yesterday, today, and tomorrow. Front Pharmacol. 2024;15: 1399121. CrossRef PubMed Google Scholar
-
3.Zheng Q, Wang X. Alzheimer's disease: insights into pathology, molecular mechanisms, and therapy. Protein Cell. 2025;16(2): 83-120. CrossRef PubMed Google Scholar
-
4.Li Z, Zhang Z, Yu B. Unlocking the therapeutic potential of natural products for Alzheimer's disease. J Med Chem. 2025;68(3): 2377-402. CrossRef PubMed Google Scholar
-
5.Committee of Flora Reipublicae Popularis Sinicae. Flora Reipublicae Popularis Sinicae. vol. 40. Beijing: China Science Press, 1994: 36. PubMed Google Scholar
-
6.Li WR, Li QG. Dictionary of Traditional Chinese Medicine Nicknames. Beijing: China Science and Technology Press, 1994: 441. PubMed Google Scholar
-
7.Wang GQ. National Chinese Herbal Medicine Compilation. vol. 4. Beijing: People's Medical Publishing House, 2005: 579. PubMed Google Scholar
-
8.Lu Y, Zhu S, He Y, Peng C, Wang Z, Tang Q. Phytochemical profile and antidepressant effect of Ormosia henryi Prain leaf ethanol extract. Int J Mol Sci. 2019;20: 3396. CrossRef PubMed Google Scholar
-
9.Feng S, Hao J, Xu Z, Chen T, Qiu SX. Polyprenylated isoflavanone and isoflavonoids from Ormosia henryi and their cytotoxicity and anti-oxidation activity. Fitoterapia. 2012;83: 161-5. CrossRef PubMed Google Scholar
-
10.Kinghorn AD, Hussain RA, Robbins EF, Balandrin MF, Stirton CH, Evans SV. Alkaloid distribution in seeds of Ormosia, Pericopsis and Haplormosia. Phytochemistry. 1988;27(2): 439-44. CrossRef PubMed Google Scholar
-
11.Kennelly EJ, Flynn TJ, Mazzola EP, Roach JA, McCloud TG, Danford DE, Betz JM. Detecting potential teratogenic alkaloids from blue cohosh rhizomes using an in vitro rat embryo culture. J Nat Prod. 1999;62(10): 1385-9. CrossRef PubMed Google Scholar
-
12.Arriola K, Guarino S, Schlawis C, Arif MA, Colazza S, Peri E, Schulz S, Millar JG. Identification of brassicadiene, a diterpene hydrocarbon attractive to the invasive stink bug Bagrada hilaris, from volatiles of cauliflower seedlings, Brassica oleracea var. botrytis. Org Lett. 2020;22(8): 2972-5. CrossRef PubMed Google Scholar
-
13.Flack HD, Bernardinelli G. Absolute structure and absolute configuration. Acta Cryst. 1999;A55: 908–15. PubMed Google Scholar
-
14.Ricker M, Daly DC, Veen EG, Robbins EF, Sinta VM, Chota IJ, Czygan F-C, Kinghorn AD. Distribution of quinolizidine alkaloid types in nine Ormosia species (Leguminosae-Papilionoideae). Brittonia. 1999;51(1): 34-43. CrossRef PubMed Google Scholar
-
15.Scharnagel D, Goller J, Deibl N, Milius W, Breuning M. The enantioselective total synthesis of bisquinolizidine alkaloids: a modular "inside-out" approach. Angew Chem Int Ed. 2018;57(9): 2432-5. CrossRef PubMed Google Scholar
-
16.Mclean S, Lau PK, Cheng SK, Murray DG. Alkaloids of certain oriental Ormosia species. Can J Chem. 1971;49(11): 1976-8. CrossRef PubMed Google Scholar
-
17.Zhou QQ, Xie XY, Zhu JW, Pan WW, Xie BG, Zhou W, Xu JB. Hosimosines A-E, structurally diverse cytisine derivatives from the seeds of Ormosia hosiei Hemsl. et Wils. Fitoterapia. 2023;170: 105661. CrossRef PubMed Google Scholar
-
18.Le PM, Martin MT, Van Hung N, Guénard D, Sévenet T, Platzer N. NMR study of quinolizidine alkaloids: relative configurations, conformations. Magn Reson Chem. 2005;43(4): 283-93. CrossRef PubMed Google Scholar
-
19.Galasso V, Przybył AK, Christov V, Kovač B, Asaro F, Zangrando E. Theoretical and experimental studies on the molecular and electronic structures of cytisine and unsaturated keto-sparteines. Chem Phys. 2006;325(2–3): 365-77. CrossRef PubMed Google Scholar
-
20.Abdel-Halim OB, Sekine T, Saito K, Halim AF, Abdel-Fattah H, Murakoshi I. (+)-12α-hydroxylupanine, a lupin alkaloid from Lygos raetam. Phytochemistry. 1992;31(9): 3251-3. CrossRef PubMed Google Scholar
-
21.Przybył AK, Kubicki M. Simple and highly efficient preparation and characterization of (−)-lupanine and (+)-sparteine. Tetrahedron. 2011;67(40): 7787-93. CrossRef PubMed Google Scholar
-
22.Khakimova TV, Pukhlyakova OA, Shavaleeva GA, Fatykhov AA, Vasil'eva EV, Spirikhin LV. Synthesis and stereochemistry of new N-substituted cytisine derivatives. Chem Nat Compd. 2001;37: 356-60. CrossRef PubMed Google Scholar
-
23.Chen H, Zhu Y, Zhang YL, Zeng MN, Cao YG, Sun PT, Cao B, Du K, Zhao X, Wang XW, Zheng XK, Feng WS. Neolignans and amide alkaloids from the stems of Piper kadsura and their neuroprotective activity. Phytochemistry. 2022;203: 113336. CrossRef PubMed Google Scholar
-
24.Zhang LJ, Zhou WJ, Ni L, Huang MQ, Zhang XQ, Xu HY. A review on chemical constituents and pharmacological activities of Ormosia. Chin Tradit Herbal Drugs. 2021;52(14): 4433-42. PubMed Google Scholar
-
25.Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J Appl Crystallogr. 2009;42: 339-41. CrossRef PubMed Google Scholar
-
26.Sheldrick GM. SHELXT–integrated space-group and crystal structure determination. Acta Cryst. 2015;A71: 3–8. PubMed Google Scholar
-
27.Yu MY, Liu SN, Luo EE, Jin Q, Liu H, Liu HY, Luo XD, Qin XJ. Phloroglucinols with hAChE and α-glucosidase inhibitory activities from the leaves of tropic Rhodomyrtus tomentosa. Phytochemistry. 2022;203: 113394. CrossRef PubMed Google Scholar
-
28.Yu MY, Liu SN, Liu H, Meng QH, Qin XJ, Liu HY. Acylphloroglucinol trimers from Callistemon salignus seeds: Isolation, configurational assignment, hAChE inhibitory effects, and molecular docking studies. Bioorg Chem. 2021;117: 105404. CrossRef PubMed Google Scholar
Copyright information
© The Author(s) 2025.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
