


Xylariaides A and B, novel cytochalasans with a unique 5/6/5/3 ring system from a soil fungus Xylaria sp. Y01
Abstract
Two new cytochalasans, xylariaides A (1) and B (2), were isolated and identified from a soil fungus Xylaria sp. Y01. Their structures were unambiguously determined by extensive spectroscopic methods including high resolution electrospray ionization mass spectrometry, ultraviolet radiation, infrared spectroscopy, and 1D/2D NMR, as well as in-depth quantum chemical calculations of gauge-including atomic orbital (GIAO) 13C NMR chemical shifts, electronic circular dichroism (ECD), and spin–spin coupling constants. The unprecedented core structure with a 5/6/5/3 fused tetracyclic ring system further enriches the scaffold types of cytochalasans.Graphical Abstract

Keywords
Cytochalasins Xylaria sp. Coupling constant calculation ECD calculation GIAO 13C NMR calculation1 Introduction
The fungal genus Xylaria, a member of the Xylariaceae family, is widely distributed in both marine and terrestrial habitats [1]. Most Xylaria species are saprophytic fungi that decompose decaying wood, bark, dung, and other organic resources [1, 2]. The capacity of produceing complex, unique, and potent bioactive secondary metabolites is their well-known characteristic. As of 2020, 245 compounds (118 new ones) with a variety of bioactivities have been discovered from Xylaria species, including terpenoids, alkaloids, polyketides, cytochalasans, aromatic compounds, and sesquiterpenoids [1]. Many of them have shown tremendous potential to be developed as lead compounds [3].
Cytochalasans are a group of fungal secondary metabolites derived from hybrid pathways of polyketide synthase–non-ribosomal peptide synthetase (PKS–NRPS) with certain amino acids, typically with a tricyclic core scaffold formed by intramolecular Diels-Alder (IMDA) cyclization [4]. In recent years, there has been a surge of novel cytochalasans from Xylaria species, including newly discovered carbon skeleton types possessing 5/6/6/6 tetracyclic ring system (curtachalasin A) [5], bridged 6/6/6/6 ring system (curtachalasin C) [6], 6/7/5/6/6/6 polycyclic system (xylarichalasin A) [7], and open-chain merocytochalasans (perochalasins A−C) [8]. Moreover, these compounds also show a broad range of bioactivities, placing them at the forefront of research in natural medicinal chemistry [9]. Attracted by the active metabolites of soil-inhabiting fungi [10], we have conducted continuous research [11, 12] and identified a strain of fungus, Xylaria sp. Y01, from which a series of cytochalasans with cytotoxic effects were discovered [13]. In subsequent in-depth studies, we further discovered two new cytochalasans (1 and 2) with an unprecedented skeleton (Fig. 1). Herein, we report their isolation, structural elucidation, and preliminary bioactivity screening results.
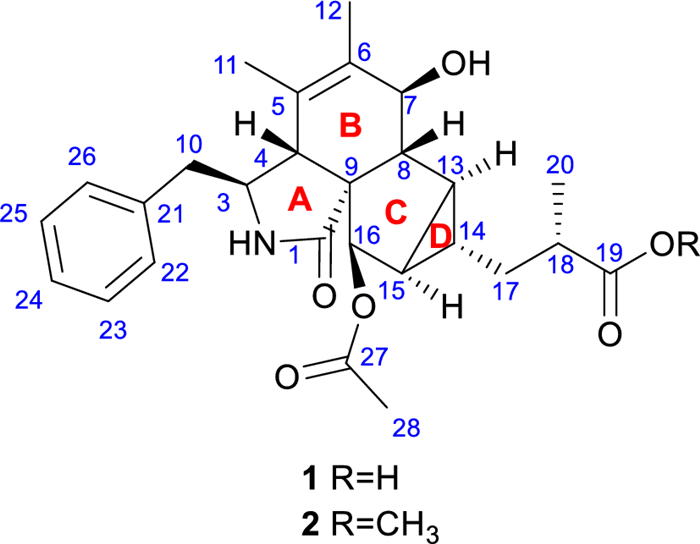
The structures of compounds 1 and 2
2 Results and discussion
2.1 Planar configuration elucidation
Compound 1 was obtained as a yellow oil with the molecular formula C27H33NO6 determined by HRESIMS at m/z 468.2383 [M + H]+ (calcd. for C27H34NO6+, 468.2381), corresponding to 12 degrees of unsaturation. The infrared absorption spectrum (IR) peaks at 3443.9, 1683.3, and 1628.6 cm−1 correspond to the stretching vibrations of the hydroxyl, carboxylic carbonyl, and amide carbonyl groups, respectively. Its ultraviolet (UV) spectrum exhibited an absorption maximum at 257 nm, indicating the existence of conjugated systems.
The 1H NMR data (Table 1) revealed characteristic signals for a monosubstituted phenyl at δH 7.11 (2H, d, J = 7.6 Hz, H-22/H-26), 7.31 (2H, t, J = 7.6 Hz, H-23/H-25), and 7.24 (1H, t, J = 7.6 Hz, H-24), three singlet methyl groups at δH 1.40 (3H, s, H3-11), 1.64 (3H, s, H3-12), and 2.17 (3H, s, H3-28), a doublet methyl group at δH 1.16 (3H, d, J = 7.0 Hz, H3-20), and one amide NH proton at δH 6.09. The spectroscopic data also indicated two distinct groups of isolated and geminally coupled protons, one methylene at δH 2.61 (1H, dd, J = 13.3, 7.5 Hz) and 2.76 (1H, dd, J = 13.3, 7.8 Hz), the other at δH 1.65 (1H, m) and 1.47 (1H, ddd, J = 14.4, 8.7, 6.0 Hz). The 13C NMR, DEPT, and HSQC data of 1 displayed 27 carbon signals, including a carboxylic acid carbon at δC 179.6 (C-19), an amide carbonyl at δC 177.4 (C-1), an ester carbonyl at δC 170.0 (C-27), a double bond at δC 127.3 (C-5) and 134.8 (C-6), one nonprotonated carbon at δC 63.3 (C-9), two methylene carbons at δC 36.3 (C-17) and 42.7 (C-10), nine sp3 methine carbons at δC 28.9 (C-13), 29.8 (C-14), 32.0 (C-15), 38.6 (C-18), 47.5 (C-4), 52.4 (C-8), 58.1 (C-3), 70.8 (C-7), and 78.1 (C-16), two of which are oxygenated. These findings suggest that compound 1 contains four double bonds and one phenyl group, inferring the presence of four rings based on the 12 degrees of unsaturation derived from the molecular formula.
The 1H NMR data (600 MHz) and 13C NMR data (150 MHz) of compounds 1 and 2 in CDCl3 (δ in ppm)
The 1H–1H COSY spectrum of 1 indicated three proton spin–spin systems, namely H2-10/H-3/H-4/2-NH, H-7/H-8/H-13/H-14(/H-15/H-16)/H2-17/H-18/H3-20, and H-22/H-23/H-24/H-25/H-26 (Fig. 2). The HMBC correlations (Fig. 2) between NH and C-1/C-3/C-4/C-9, H-4 and C-1/C-6/C-10, H-7 and C-5, H3-11 and C-4/C-5/C-6, H3-12 and C-5/C-6/C-7, as well as H-8 and C-4/C-6 established the structural connectivity between the five-membered lactam ring (A ring) and the six-membered ring (B ring). A proposed ring C fused with a tricyclic ring D was deduced by the HMBC correlations between H-13 and C-7/C-16/C-17, H-15 and C-9/C-8/C-17, H-16 and C-1/C-8/C-14, as well as H-14 and C-8/C-16, along with the 1H–1H COSY correlations of H-13/H-15/H-14. Additionally, the spin–spin coupling of H-14/H2-17/H-18/H3-20, and the HMBC correlations between H3-20 and C-17/C-18/C-19, H2-17 and C-13/C-15/C-19 suggested the presence of a 2-methylpropanoic acid moiety attached to C-14 of the tricyclic ring (D ring). Further HMBC correlation between H-16 and C-27 verified the attachment of the acetyl group at C-16. Thus, the planar structure of compound 1 was defined as a novel cytochalasan skeleton with a unique bicyclo[3.1.0]hexane moiety.
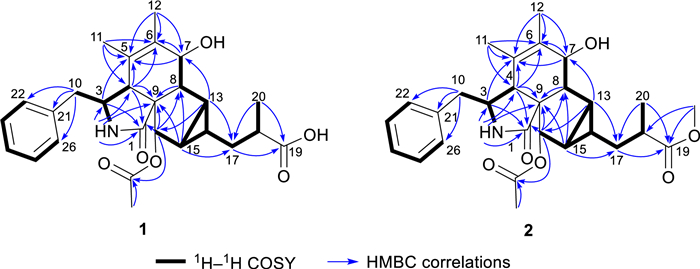
1H–.1H COSY and key HMBC correlations of compounds 1 and 2
Nuclear Overhauser effects (NOEs) between H-4 and H-16, as well as between H2-10 and H-16, suggest that these protons are on the same side of the five-membered lactam ring. The NOESY spectrum showed correlations of H-4/H-8/H-14 and of H-7/H-13, suggesting that H-4, H-8, and H-14 are cofacial, while H-7 and H-13 are cofacial (Fig. 3). The equatorial position of H-7 was inferred from the coupling constant between H-7 and H-8 (J = 10.5 Hz) (Table 1). Previous studies have indicated that the essential elements of most cytochalasan skeletons share the same stereochemistry, with configurations at C-3, C-4, C-7, C-8, C-9, and C-16 assigned as 3S, 4R, 7S, 8R, 9R, and 16R, respectively [5, 13], which is consistent with the aforementioned NOE analyses. However, the relative configuration of ring D could not be fully elucidated by NOESY experiments. Fortunately, coupling constants between vicinal protons in ring D provide valuable information for determining their relative orientations. H-13 exhibited coupling constants of 7.7, 4.2, and 3.8 Hz with H-15, H-8, and H-14, respectively. The small coupling constant between H-13 and H-14 (J = 3.8 Hz) and the large coupling constant between H-13 and H-15 (J = 7.7 Hz) suggest that H-13 and H-15 are coplanar and both diaxial to H-14.
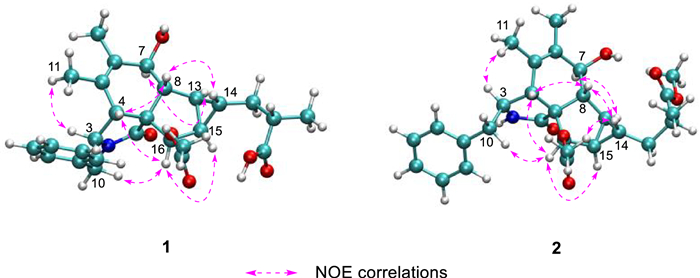
Key NOE correlations of compounds 1 and 2, depicted with their optimal conformations (coordinates are provided in Tables S14 and S22)
To confirm the relative configuration of ring D, the J coupling constants for different possible conformations of ring D were calculated [14]. The fusion of rings C and D is restricted to a cis configuration due to strain that inhibits the trans configuration from achieving a stable geometric structure. Therefore, the relative configuration of 1 is one of the eight possible relative diastereoisomers (1a–1h), as shown in Fig. 4. The J coupling constants of protons on rigid rings for these eight possible epimers were correlated with the experimental coupling constants to calculate the mean absolute error (MAE) and root mean square error (RMS). The results indicate that the relative configurations of 1d and 1h align more closely with the experimental values, as they exhibit lower MAE and RMS compared to the other epimers (Table 2). These findings are consistent with the previous analysis.

Eight possible epimers of compounds 1 and 2
Experimental J coupling constants (CDCl3) for compounds 1 and 2, and theoretical values for eight possible epimers (detailed data are provided in Table S5 and S6; the values in bold font are the optimal values in their respective columns)
To discern the chirality at key positions from an alternative perspective, 13C NMR calculations were utilized by analyzing chemical shift differences. The GFN2NMR method, a semiempirical approach integrated into a deep graph convolutional network, facilitates rapid and precise 13C NMR chemical shift calculations, noted for its high accuracy and low computational cost [15]. It is capable of distinguishing diastereomers based on the rules learned from large datasets. The most likely structures of 1 were swiftly identified from eight possible isomers using the GFN2NMR method. Since their Pmean values fall within the confidence interval and Prel exceeds 5% (1d at 71%, 1h at 29%), both 1d and 1h are assigned as plausible configurations (Table 3).
Experimental 13C NMR chemical shifts (CDCl3) of compounds 1 and 2, and the calculated chemical shifts for the eight possible epimers predicted by GFN2NMR (detailed data are provided in Table S1 and S2; the values in bold font are the optimal values in their respective columns)
The GIAO 13C NMR calculations with the sorted training sets (STS) protocol showed significantly enhanced accuracy and reliability for structural determination [16] (e. g., pestalopyrones A–D [12]), which provided a tool based on quantum chemistry. The calculated chemical shifts by STS for the eight possible epimers 1a–1h were correlated with the experimental 13C NMR chemical shifts to determine MAE, RMS, and the statistical parameters Pmean and Prel (Table 4). The 13C NMR calculations for compound 1h closely matched the experimental data, with both the MAE and RMS values significantly lower than those for 1b. Furthermore, the Prel probability calculations assigned a 100% likelihood to 1h. This result not only established the chirality at C-18 but also validated the conformation of the D ring, consistent with the possible structures inferred from J coupling constants calculations and GFN2NMR analysis. An ECD calculation for 1h was conducted to determine the absolute configuration of 1, indicating a configuration of 3S,4R,7S,8R,9R,13S,14S,15R,16R,18S, which matched well with the experimental ECD data as shown in Fig. 5. These findings confirm that 1h is the correct structure of xylariaide A.
13C NMR chemical shifts calculation (CDCl3) of structures a–h fitting to the experimental data of compounds 1 and 2 using GIAO 13C NMR calculations with the STS protocol (detailed data are provided in Table S3 and S4; the values in bold font are the optimal values in their respective columns)
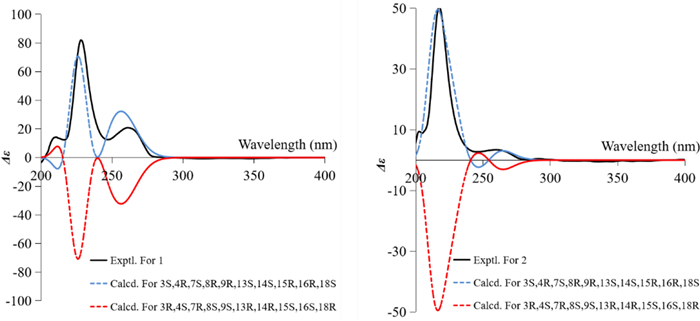
The experimental and calculated ECD curves of 1 and 2 and their enantiomers
Compound 2 was obtained as an amorphous powder, with a molecular formula of C28H35NO6, as determined by HRESIMS at m/z 482.2538 [M + H]+ (calcd. for C28H36NO6+, 482.2538). The one-dimensional spectrum data of compound 2 exhibited an additional methoxyl group at δH 3.68 (3H, s) and δC 51.7, with all other signals closely resembling those of 1 except for the active proton signal of 2-NH (Table 1). The HMBC correlation between -OCH3 and C-19 confirmed the attachment of the methoxyl group at C-19. NOE signals indicate that H-4, H2-10, and H-16 are on the same side of the five-membered lactam ring, while H3-11 and H-3 are on the opposite side. The optimal conformations of compounds 1 and 2, as illustrated in Fig. 3, differ significantly in the configuration of the 2-methylpropionic acid moiety, leading to the absence of a distinct NOE signal between H-14 and H-18 for 2. The remaining NOE signals for 2 are consistent with those of 1. Similarly, the J coupling constants and 13C NMR calculations of 2 were also performed, aligning with the results for 1 and confirming the relative configuration of 2. The experimental ECD spectrum of 2 displayed two positive Cotton effects at 215 nm (Δε + 52.6) and 258 nm (Δε + 3.5), similar to the spectrum of 1. Absolute confirmation was determined by ECD calculation, with the experimental values closely matching those for the configuration 3S,4R,7S,8R,9R,13S,14S,15R,16R,18S, namely xylariaide B.
Upon comparing the structures of compounds 1 and 2, it is evident that their only difference lies in the methoxy substitution on the 19-COOH group at the side chain. In the 1D NMR spectra, most of their signals are remarkably similar. However, the chemical shift of the 2-NH proton, which is distant from the side chain, shows a significant difference of 0.6 ppm. To investigate the cause of this discrepancy and further validate the structural assignments, we analyzed the low-energy conformations of compounds 1 and 2, as shown in Fig. 6. It revealed that the most dominant conformation of compound 1 features the 10-phenyl ring and the N-2 group adopting an anti-conformation. In contrast, for compound 2, the gauche conformation is energetically more favorable and predominant. In this conformation, the 2-NH proton lies within the shielding region of the 10-phenyl ring, which explains why the chemical shift of 2-NH in compound 2 appears at a higher field in the 1H NMR spectrum. Further examination of their geometric structures revealed that in the lowest-energy conformation of compound 1, a weak hydrogen bond (bond length: 2.28 Å) forms between the 19-COOH and 27-CO groups. This interaction pulls the 16-acetyl group away from the 10-phenyl ring (the distance between 22-H and C-27 is 2.79 Å, and the dihedral angle between C-27 and H-16 is 33.5º), allowing the phenyl ring to adopt the energetically favorable anti-conformation. In contrast, in the anti-conformation of compound 2, the 16-acetyl group is closer to the 10-phenyl ring (the distance between 22-H and C-27 is 2.66 Å, and the dihedral angle between C-27 and H-16 is 22.3º). The resulting steric hindrance likely increases the energy of the anti-conformation, making the gauche conformation the lowest-energy state. This analysis not only provides a clear explanation for why a simple methoxy substitution on the 19-COOH group can lead to a significant change in the chemical shift of the distant 2-NH proton, but also further corroborates the validity of the structural assignments.
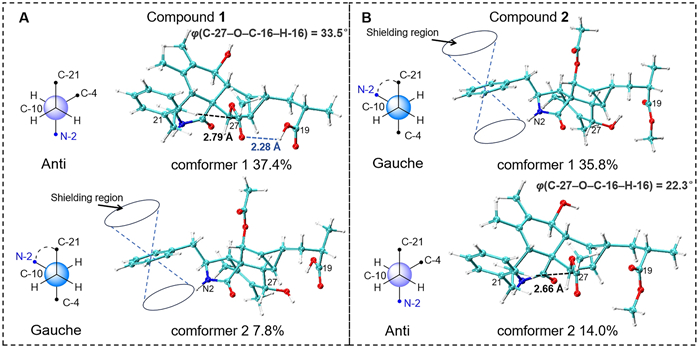
Analysis of the magnetic anisotropic effect of the benzene ring on the 2-NH proton in compounds 1 and 2. Conformer 1 of compounds 1 and 2 represents the global minimum conformation, respectively
2.2 Proposed biosynthetic pathway
Compounds 1 and 2 contain the same rings A and B as curtachalasin J [17], a co-isolated known cytochalasan identified from this strain (NMR data provided in Figs. S22 and S23), and also feature a benzyl group at C-3. This observation suggests that the 5/6/5/3 ring system common to compounds 1 and 2 may be biogenetically derived from the known compound curtachalasin J. Intramolecular cyclization between C-13 and C-20 in curtachalasin J could result in the bicyclo[3.1.0]hexane moiety. Besides, inspired by the C–C bond cleavage reactions on α-hydroxyketones [18], we propose that the carbon bonds between C-17 and C-18 and between C-17 and C-19 within ring E undergo oxidative cleavage, and both C-17 and C-19 are transformed into carboxylic acids. The following decarboxylation at C-19 leads to the formation of compound 1. The stereochemistry of C-8, C-13, C-14, C-16, C-20, and C-21 in curtachalasin J corresponds to that of C-8, C-13, C-14, C-18, C-15, and C-16 in compound 1, respectively, further supporting the proposed biogenetic pathway (Scheme 1).
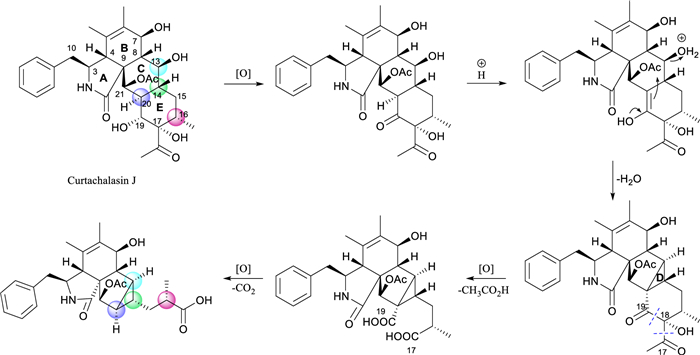
Proposed Biosynthetic Pathway of compounds 1 and 2
2.3 Preliminary bioactivity screening results
The cytochalasans, a well-known family of fungal metabolites, are recognized for their ability to inhibit the polymerization of filamentous (F)-actin [19]. These compounds have shown significant anticancer properties in numerous in vitro and in vivo studies [20]. To evaluate the potential anticancer properties of compounds 1 and 2, a cytotoxicity assessment was conducted on three cancer cell lines (HCT116, 4T1, and MHCC97H) and one normal cell line (GES-1). Compounds 1 and 2 did not exhibit significant cytotoxic effects against any of the cell lines tested.
To assess the antimicrobial activity of compounds 1 and 2, a screen for antibacterial activity against Bacillus subtilis, Escherichia coli, Staphylococcus aureus, Acinetobacter baumannii, and Pseudomonas aeruginosa, as well as for antifungal activity against Saccharomyces cerevisiae and Canidia albicans, was conducted. Both compounds showed MIC values above 50 μM, indicating no significant antimicrobial activity against the bacterial strains tested.
The anti-inflammatory activity of compounds 1 and 2 was evaluated by assessing their inhibitory effects on nitric oxide (NO) production in lipopolysaccharide (LPS)-stimulated RAW264.7 macrophages. Compounds 1 and 2 demonstrated no significant inhibitory activity on NO production at a concentration of 30 μM.
3 Experimental
3.1 General
UV spectra were measured by Nano-300 Microvolume Spectrophotometer (Hangzhou Allsheng Instruments) with a 1.0 mm pathlength. CD spectra were recorded using a ChirascanTM-plus Circular Dichroism spectrometer (Applied Photophysics Ltd.). Optical rotations were measured using a Autopol Ⅳ automatic polarimeter (Rudolph Research Analytical). IR spectra were recorded on a Shimadzu Fourier Transform Infrared Spectrometer using KBr pellets. HRESIMS spectra were obtained with an Agilent 6500 series Q-TOF mass spectrometer (Agilent Technologies, Singapore). NMR spectra were obtained at 600 MHz for 1H and 150 MHz for 13C on Bruker spectrometers (Bruker BioSpin, Rheinstetten, Germany). Column chromatography (CC) was performed with macroporous adsorbent resin D101 (Tianjin Haoju Resin Technology Co., LTD, Tianjin, China), and silica gel (200 − 300 mesh and 300 − 400 mesh, Qingdao Marine Chemical, Qingdao, China). Thin Layer Chromatography (TLC) separations were executed on precoated silica gel GF254 plates (Qingdao Marine Chemical), with visualization under UV light or by spraying with 5–15% H2SO4 in EtOH followed by heating. HPLC separation was performed using a Ruihe Tech prep-HPLC system (xgt6515-10 s) with SinoChrom ODS-AP (Elite, 5 μm, 10 × 250 mm) and a DAD detector (Ruihe tech, LC2050).
3.2 Fungi culture, extraction and isolation
A soil sample from Yunnan, China, yielded a strain of Xylaria designated as Y01. The methods for sample processing and fungal species identification followed those outlined in our previous publication [13]. Xylaria sp. Y01 hyphae were cultivated in SDA liquid medium at a constant temperature of 25 ℃ for 10 days. These cultures were then transferred to rice medium in 500 mL Erlenmeyer flasks and incubated at 25 ℃ for 40 days. A total of 10 kg of rice was used for the cultures. The fermented rice medium was extracted with 30 L of ethyl acetate, repeated ten times at room temperature, yielding a crude extract of 200 g. This extract was processed through 300 g of macroporous resin D101, followed by sequential elution with water, 80% aqueous methanol, and pure methanol. The fraction eluted with the 80% methanolic solution was collected and concentrated to yield a residue of 100 g. This methanol eluate between 80 and 90% was further subjected to column chromatography on silica gel, using a gradient of petroleum ether and ethyl acetate in ratios ranging from 5:1 to 0:1, to separate it into five distinct subfractions (Fr. A to E). The remaining Fr. B was purified by a semipreparative HPLC system successively to yield 1 (5.11 mg, 25% methanol, 3.0 mL/min, tR = 25.0 min, SinoChrom ODS-AP: 10.0 mm × 250 mm, 5 μm), and 2 (0.45 mg, 60% methanol, 2.5 mL/min, tR = 14.0 min, SinoChrom ODS-AP: 10.0 mm × 250 mm, 5 μm).
3.3 Compound characterization
Xylariaide A (1): Yellow oily. [α]D25 + 52.00 (c 0.511, MeOH). IR (KBr): υmax 3443, 1683, 1628, 1379.9, 1125, 854 cm−1. UV (CH3CN) λmax (log ε) 257 (2.5) nm. 1H NMR (600 MHz in CDCl3) and 13C NMR (150 MHz in CDCl3) data, see Table 1. HRESIMS m/z 468.2383 [M + H]+ (calcd. for C27H34NO6+, 468.2381, Δ + 0.6 ppm), m/z 450.2279 [M + H − H2O]+ (calcd. for C27H32NO5+, 450.2275, Δ + 0.9 ppm).
Xylariaide B (2): White powder. [α]D25 + 142.00 (c 0.066, MeOH). UV (CH3CN) λmax (log ε) 257 (2.4) nm. 1H NMR (600 MHz in CDCl3) and 13C NMR (150 MHz in CDCl3) data, see Table 1. HRESIMS m/z 482.2538 [M + H]+ (calcd. for C28H36NO6+, 482.2538, Δ 0.0 ppm), m/z 464.2431 [M + H − H2O]+ (calcd. for C28H34NO5+, 464.2432, Δ − 0.2 ppm). The IR spectrum was not measured because of its low sample amount.
3.4 Assay of antimicrobial activity
The antimicrobial activity of the two cytochalasans (1 and 2) against B. subtilis ATCC 6051, E. coli DH5α, S. aureus ATCC25923, A. baumannii ATCC19606, and P. aeruginosa ATCC27853 as well as S. cerevisiae ATCC9763 and Canidia albicans ATCC10231 was evaluated. Samples were prepared by dissolving them in an 80% ethanol aqueous solution to achieve an initial concentration of 1 mM. The samples were then diluted to a final concentration of 7.5 × 105 CFU/mL, and bacterial solutions were added to each well of a 96-well culture plate. The plate was incubated at 37 ℃ for 24 h, after which the wells were visually inspected to evaluate growth inhibition or clearing of the bacterial cultures. The experiment included media blank, bacterial control, streptomycin-positive bacterial control, and fluconazole-positive fungal controls for comparative analysis.
3.5 Assay of cytotoxic activity
The cell culture and cytotoxic activity assays were conducted as previously described [13].
3.6 Assay of anti-inflammatory activity
RAW264.7 cells were cultured in DMEM medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 1 mM sodium pyruvate, and 2 mg/mL gentamicin, and maintained in a humidified atmosphere at 37 ℃ with 5% CO2. For the assay, cells were seeded into 96-well plates at a density of 5 × 105 cells per well and incubated with the two isolates (1 and 2) at a concentration of 30.0 μM for an additional 24 h under the same conditions. After 24 h of incubation, 10 μL of CCK-8 reagent was added to the cell medium and incubated at 37 ℃ for 4 h. The absorbance was measured at 450 nm using a microplate reader. Absorbance values from untreated cells were taken as 100% viability. No significant cytotoxic effect was observed, permitting the use of concentrations below 30 μM of 1 and 2 in subsequent experiments.
Nitric oxide (NO) production was determined by measuring the nitrite content in the cell culture supernatant using Griess reagent. RAW264.7 cells were treated with LPS (1 µg/mL) in the presence or absence of 1 and 2 (30 µM) for 24 h. 100 µL of the cell culture supernatant was mixed with 100 µL of Griess reagent and incubated for 10 min at room temperature. The absorbance at 540 nm was measured.
3.7 Quantum chemical calculation
The software Crest [21] was employed to explore the conformational space of potential structures within the GFN0 [22, 23] theoretical framework. Subsequently, structures were optimized at the GFN2-xTB [22] level, with a threshold of 4 kcal/mol to exclude high-energy conformers. Optimization and frequency calculation of each conformer were performed on B3LYP-D3(BJ)/TZVP (IEFPCM) level of theory. DFT GIAO 13C NMR calculations were performed on the ωB97x-D/6-31G* (IEFPCM, chloroform) level, aligning with the reported STS protocol [16]. ECD calculations for 1 and 2 were executed on the B3LYP/TZVP (IEFPCM, methanol) level. The calculated shielding tensors and ECD curves of conformers were Boltzmann-averaged based on Gibbs free energy. SpecDis V1.71 [24] was used to Boltzmann-average and simulate the ECD curves of all conformers. For both 1 and 2, sigma/gamma values were set to 0.3 eV, and the calculated curves were red-shifted by 25 nm. The J coupling constants calculations for protons in rigid rings were conducted using B3LYP/PCSSEG-2 (CPCM, chloroform) with the ORCA program [25]. GIAO 13C NMR and ECD calculations were executed using the Gaussian 16 software package [26]. Supplementary material includes DFT-optimized geometry data, relative energies, and conformational populations of all calculated structures. The 3D structures of 1 and 2 were generated using VMD (1.9.4) [27].
4 Conclusions
This study reports the discovery of xylariaides A and B, which feature a novel cytochalasan skeleton. Although efforts to obtain single crystals for X-ray diffraction analysis were unsuccessful, the structure was reliably determined using a combination of spectroscopic methods, 13C NMR calculations with the STS protocol, and ECD calculations. These three distinct validation techniques each effectively confirmed the conformation of the tricyclic ring system. Despite the lack of positive results from recent bioactivity assays, these findings enrich the scaffolds of cytochalasans and lay a foundation for future research.
Notes
Acknowledgements
The authors thank the Institute for Advanced Study, Central South University for NMR measurement.
Author contributions
Yi-Yun Yuan: Writing–original draft, Methodology, Investigation. Yan Li: Investigation. Wen-Yu Lu: Methodology. Ai-Lin Liang: Methodology. Jing Li: Writing–review & editing, Investigation, Funding acquisition. Wen-Xuan Wang: Writing–review & editing, Writing–original draft, Funding acquisition, Conceptualization.
Funding
This work was supported by National Natural Science Foundation of China (82173713), Key Research and Development Program of Hunan Province (2022SK2031), and Natural Science Foundation of Hunan Province (2024JJ8127). Project of Hunan Administration of Traditional Chinese Medicine (B2023059), Changsha Municipal Natural Science Foundation (kq2403041).
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Declarations
Competing interests
The authors declare no conflict of interest in this manuscript.
References
-
1.Chen W, Yu M, Chen S, Gong T, Xie L, Liu J, Bian C, Huang G, Zheng C. Structures and biological activities of secondary metabolites from Xylaria spp. J Fungi. 2024;10(3): 90. CrossRef PubMed Google Scholar
-
2.Song F, Wu SH, Zhai YZ, Xuan QC, Wang T. Secondary metabolites from the genus Xylaria and their bioactivities. Chem Biodivers. 2014;11(5): 673-94. CrossRef PubMed Google Scholar
-
3.Macías-Rubalcava ML, Sánchez-Fernández RE. Secondary metabolites of endophytic Xylaria species with potential applications in medicine and agriculture. World J Microbiol Biotechnol. 2016;33(1): 15. CrossRef PubMed Google Scholar
-
4.Scherlach K, Boettger D, Remme N, Hertweck C. The chemistry and biology of cytochalasans. Nat Prod Rep. 2010;27(6): 869-86. CrossRef PubMed Google Scholar
-
5.Wang WX, Li ZH, Feng T, Li J, Sun H, Huang R, Yuan QX, Ai HL, Liu JK. Curtachalasins A and B, two cytochalasans with a tetracyclic skeleton from the endophytic fungus Xylaria curta E10. Org Lett. 2018;20(24): 7758-27761. CrossRef PubMed Google Scholar
-
6.Wang WX, Lei X, Ai HL, Bai X, Li J, He J, Li ZH, Zheng YS, Feng T, Liu JK. Cytochalasans from the endophytic fungus Xylaria cf curta with resistance reversal activity against fluconazole-resistant candida albicans. Org Lett. 2019;21(4): 1108-11. CrossRef PubMed Google Scholar
-
7.Wang WX, Lei X, Yang YL, Li ZH, Ai HL, Li Jing, Feng Tao, Liu Ji-Kai. Xylarichalasin a halogenated hexacyclic cytochalasan from the Fungus Xylaria cf curta. Org Lett. 2019;21(17): 6957-60. CrossRef PubMed Google Scholar
-
8.de Amorim MR, Schoellhorn SM, Barbosa CdS, Mendes GR, Macedo KdL, Ferreira AG, Venâncio T, Guido RVC, Batista ANL, Batista JM, et al. Structure and biosynthesis of perochalasins A-C open-chain merocytochalasans produced by the marine-derived fungus Peroneutypa sp M16. J Nat Prod. 2024;87(9): 2204-15. CrossRef PubMed Google Scholar
-
9.Lambert C, Schmidt K, Karger M, Stadler M, Stradal TEB, Rottner K. Cytochalasans and their impact on actin filament remodeling. Biomolecules. 2023;13(8): 1247. CrossRef PubMed Google Scholar
-
10.Zhang D, Li S, Fan M, Zhao C. The novel compounds with biological activity derived from soil fungi in the past decade. Drug Des Devel Ther. 2022;16: 3493-555. CrossRef PubMed Google Scholar
-
11.Mo JS, Tan YF, Ai WJ, Li Y, Yuan YY, Jiang Y, Xu K, Tan G, Wang WX, Li J, et al. Macrocyclic trichothecenes from Myrothecium verrucaria PA 57 and their cytotoxic activity. Chin J Nat Med. 2024;22(9): 854-63. PubMed Google Scholar
-
12.Wu XQ, Li J, Zhou X, Wang J, Tan YF, Mo JS, Liu S, Xu KP, Tan GS, Zhang W, et al. Liver-cell protective pyridones from the fungi Tolypocladium album dws120. Phytochemistry. 2023;212: 113730. CrossRef PubMed Google Scholar
-
13.Yuan YY, Li Y, Li J, Lu WY, Liang AL, Xu PJ, Wang WX. Chemical constituents from a soil fungus Xylaria sp Y01 and their anticancer potential. J Mol Struct. 2025;1321(5): 140274. CrossRef PubMed Google Scholar
-
14.Radulović NS, Mladenović MZ, Stojanović NM, Randjelović PJ, Blagojević PD. Structural elucidation of presilphiperfolane-7α, 8α-diol, a bioactive sesquiterpenoid from pulicaria vulgaris: a combined approach of solvent-induced chemical shifts, GIAO Calculation of chemical shifts, and full spin analysis. J Nat Prod. 2019;82(7): 1874-85. CrossRef PubMed Google Scholar
-
15.Ai WJ, Li J, Cao DS, Liu S, Yuan YY, Li Y, Tan GS, Xu KP, Yu X, Kang F, et al. A very deep graph convolutional network for 13C NMR chemical shift calculations with density functional theory level performance for structure assignment. J Nat Prod. 2024;87(4): 743-52. CrossRef PubMed Google Scholar
-
16.Li J, Liu JK, Wang WX. GIAO 13C NMR calculation with sorted training sets improves accuracy and reliability for structural assignation. J Org Chem. 2020;85(17): 11350-8. CrossRef PubMed Google Scholar
-
17.Wang WX, Cheng GG, Li ZH, Ai HL, He J, Li J, Feng T, Liu JK. Curtachalasins immunosuppressive agents from the endophytic fungus Xylaria cf. curta. Org Biomol Chem. 2019;17(34): 7985-94. CrossRef PubMed Google Scholar
-
18.Lee JHZ, Coleman T, Mclean MA, Podgorski MN, Hayball EF, Stone ISJ, Bruning JB, Whelan F, Voss JJ, Sligar SG, Bell SG. Selective α-hydroxyketone formation and subsequent C-C bond cleavage by cytochrome P450 monooxygenase enzymes. ACS Catal. 2024;14(11): 8958-71. CrossRef PubMed Google Scholar
-
19.Casella JF, Flanagan MD, Lin S. Cytochalasin D inhibits actin polymerization and induces depolymerization of actin filaments formed during platelet shape change. Nature. 1981;293(5830): 302-5. CrossRef PubMed Google Scholar
-
20.Trendowski M. Using Cytochalasins to improve current chemotherapeutic approaches. Anticancer Agents Med Chem. 2015;15(3): 327-35. CrossRef PubMed Google Scholar
-
21.Pracht P, Bohle F, Grimme S. Automated exploration of the low-energy chemical space with fast quantum chemical methods. Phys Chem Chem Phys. 2020;22(14): 7169-92. CrossRef PubMed Google Scholar
-
22.Bannwarth C, Ehlert S, Grimme S. GFN2-xTB—an accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J Chem Theory Comput. 2019;15(3): 1652-71. CrossRef PubMed Google Scholar
-
23.Grimme S, Bannwarth C, Shushkov P. A robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent interactions of large molecular systems parametrized for all spd-block elements (Z = 1–86). J Chem Theory Compu. 2017;13(5): 1989-2009. CrossRef PubMed Google Scholar
-
24.Bruhn T, Schaumlöffel A, Hemberger Y, Bringmann G. SpecDis: quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality. 2013;25(4): 243-9. CrossRef PubMed Google Scholar
-
25.Neese F. Software update: the ORCA program system-Version 50. Wiley Interdiscip Rev Comput Mol Sci. 2022;12(5): e1606. CrossRef PubMed Google Scholar
-
26.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ. Gaussian 16. Wallingford: Gaussian Inc; 2016. PubMed Google Scholar
-
27.Fernandes HS, Sousa SF, Cerqueira N. VMD Store-a VMD plugin to browse, discover, and install VMD extensions. J Chem Inf Model. 2019;59(11): 4519-23. CrossRef PubMed Google Scholar
Copyright information
© The Author(s) 2025
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
