


Structurally diverse polyketides and alkaloids produced by a plant-derived fungus Penicillium canescens L1

Abstract
A series of structurally diverse polyketides (1–3), sesterterpenoids (24 and 25), and alkaloids (26–34) were isolated from the fermentation of a plant-derived fungus Penicillium canescens L1 on solid rice medium. Among these secondary metabolites, penicanesols A–G (1–7) were new structures, which were elucidated by NMR, HR-ESI-MS, ECD calculation, and X-ray diffraction. Penicanesol A (1) represented a rare dimer derived from phthalan derivatives, characterized by a 5/6/6/6/5 heteropentacyclic core. The bioassay on the NCI-H1975 cell model showed that two compounds had good cytotoxic activities, and the most significant activate compound 13 had an IC50 value of 4.24±0.13 μM, more than the positive control drug (12.99±0.13 μM).Graphical Abstract

Keywords
Endophytic fungus Penicillium canescens Polyketides Alkaloids Cytotoxic activity1 Introduction
Penicillium is a widely distributed filamentous fungus, which has great potential to produce novel metabolites with diverse biological activities. So far, a variety of structurally diverse secondary metabolites including polyketides, alkaloids, sterols, terpenoids, and macrolides have been discovered from this genus, and most of them have multiple biological activity potentials such as antibacterial, cytotoxic, anticancer, antioxidant, antifungal, and antiviral properties [1-5]. Drugs like penicillin, mevastatin, griseomycin, and cyclosporin are star molecules produced by the Penicillium genus.
The gray mold fungus Penicillium canescens is a common microorganism in the genus Penicillium, which is distributed in soil, ocean, and plant bodies. Previous studies have investigated the secondary metabolites of this strain and discovered various molecules with novel structures and diverse activities, including aromatic polyketides, terpenes, azathiones, anthraquinones, and alkaloids. Their pharmacological effects involve antibacterial, anticancer, and β-glucosidase inhibition [6-15].
Our research group had carried out the investigation on the chemical composition of the plant Croton cnidophyllus for the first time [16]. In order to continue searching for active molecules from endophytic fungi [17, 18], we studied the secondary metabolites of fungus P. canescens L1 isolated from the plant of C. cnidophyllus. As a result, a total of 34 structurally diverse secondary metabolites, such as polyketides (Fig. 1), sesterterpenoids (Fig. 1), and alkaloids (Fig. 2), were obtained from the fermentation of fungus P. canescens L1. Their structures were analyzed by NMR, HR-ESI–MS, ECD, and X-Ray single crystal diffraction data. The selected compounds were evaluated on the NCI-H1975 cell model for their cytotoxic activities. Herein, the isolation of all compounds produced by the plant-derived fungus P. canescens L1, the elucidation of new structures, as well as their cytotoxic activities against NCI-H1975 cell were described.

The structures of polyketides (1–23) and sesterterpenoids (24 and 25) from P. canescens L1
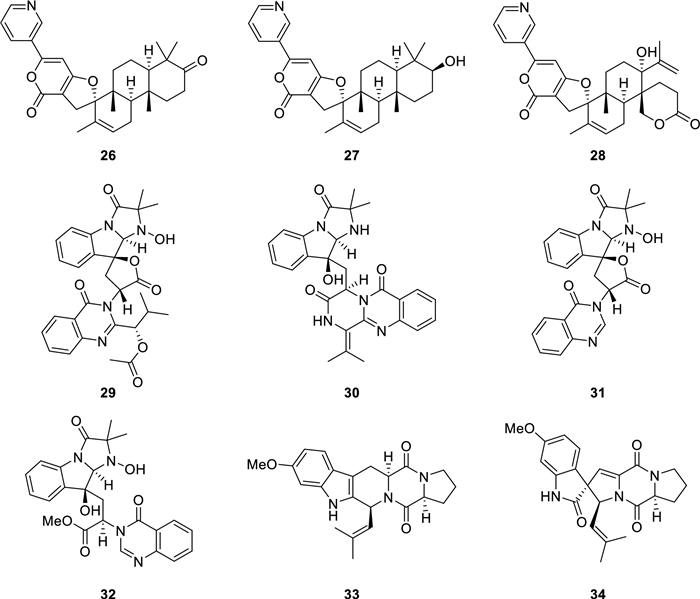
The structures of alkaloids (26–34) from P. canescens L1
2 Results and discussion
Compound 1 was obtained as colorless needles and had a molecular formula of C20H20O8 corresponding to 11 degrees of unsaturation based on HR-ESI-MS data m/z 411.1055 [M + Na]+ (calcd for C20H20O8Na+, 411.1050) and 1D NMR data. The IR spectrum of 1 exhibited absorption bands for the presence of OH (3357 cm−1), carbonyl (1705 cm−1), and phenyl (1612, 1572, and 1496 cm−1) groups. The observed signals in the 1H NMR spectrum suggested the presence of one doublet methyl [δH 1.86 (3H, d, J = 5.6 Hz)], one singlet methyl [δH 2.50 (3H, s)], two methoxy groups [δH 3.33 and 3.83 (each 3H, s)], two aromatic proton signals [δH 6.54 and 7.41 (each 1H, s)], one isolated methylene [δH 3.73 and 3.85 (each 1H, d, J = 15.8 Hz)], and a hydroxymethyl group [δH 4.95 and 5.15 (each 1H, d, J = 11.6 Hz)] (Table 1). The 13C NMR combined with DEPT spectra showed a total of 20 carbon signals (Table 1), which were classified as one conjugated carbonyl (δC 186.9), two hemiketals (δC 93.5 and 99.8), eight sp2 quaternary carbons (including four oxygenated ones), three methines (including two oxygenated ones, δC 80.4 and 137.9), two methylenes (including an oxygenated one, δC 73.5), four methyl groups including two methoxys (δC 49.7 and 56.8).
The 1H NMR (400 MHz) and 13C NMR (100 MHz) data of 1 in pyridine-d5
Analysis of the HMBC correlations, it could be determined that 1 had two similar 5/6 bicyclic units a and b. As shown in Fig. 3, the HMBC correlations from H3-8 to C-3 and C-3a, H2-1 to C-3, C-3a, C-7a, and C-7, H-7 to C-1, C-3a, and C-5, and the methoxy protons to C-6 confirmed that unit a was the same as curvulol (9) [19]. While another fragment b was also determined to have a skeleton similar to fragment a by the HMBC correlations (Fig. 3), with the difference being that its 6-membered ring was a non-aromatic ring characterized by an one conjugated carbonyl and two hemiketals and its five membered ring was a furan ring. The connection of units a and b through C-4–O–C-5′ and C-5–O–C-6′ determined by analyzing its degrees of unsaturation and comparing NMR data of similar compounds such as canescone E [14]. The relative configuration of compound 1 could not be determined by its NOE correlations. Fortunately, the crystal of 1 obtained from the solvent MeOH/DCM (5:1), and subsequent analysis of its X-ray single crystal diffraction data confirmed the structure of 1 including the (3S, 5′S, 6′S) absolute configuration (Fig. 4).

The 1H–1H COSY and key HMBC correlations of compounds 1–6
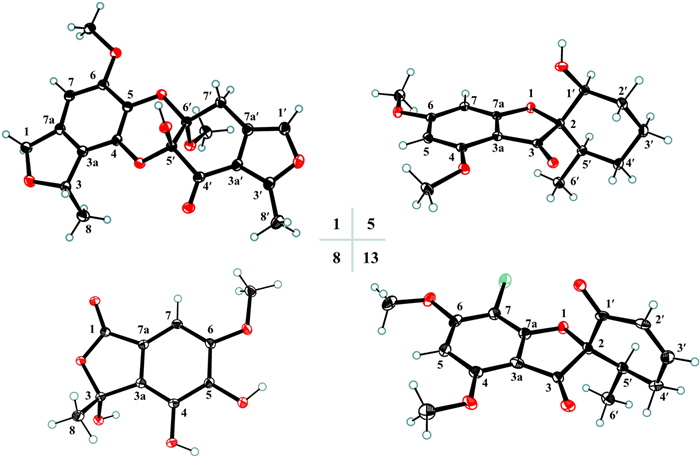
ORTEP drawings of compounds 1, 5, 8, and 13
Compound 2, a white amorphous solid, had the molecular formula C10H10O5 as determined by the HR-ESI-MS data m/z 233.0422 [M + Na]+ (calcd for C10H10O5Na+, 233.0420) and 1D NMR data. Comparison of its NMR data (Table 2) with those of 4,5,6-trihydroxy-3-methylphthalide [20] indicated that 2 possessed an additional methoxy group [δH 3.89 (3H, s)]. The HMBC correlation of this methoxy protons to C-6 confirmed its position at C-6. The absolute configuration of the only chiral carbon in compound 2 was determined by the ECD calculation method. As shown in Fig. 5, the experimental ECD curve of 2 matched well with the calculated ECD curve of (3R)-2, which indicated that the absolute configuration of C-3 in 2 was R.
The 1H NMR (400 MHz) and 13C NMR (100 MHz) data of 2–4
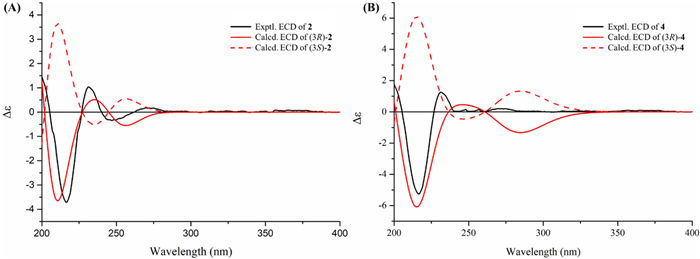
Experimental and calculated ECD spectra (A and B) of 2 and 4 in MeCN
Compound 3 was obtained as a white amorphous solid. Its molecular formula was inferred to be C11H12O5 on the basis of the HR-ESI-MS and 1D NMR data. The 1D NMR data of 3 (Table 2) was very similar to that of 2 except for the presence of an additional methoxy signal [δC 61.3; δH 3.96 (3H, s)]. This methoxy group was determined to be located at C-5 by the HMBC correlation from the methoxy protons (δH 3.96) to C-5 (δC 140.5) (Fig. 3). The absolute configuration of 3 was determined by comparison of its experimental ECD spectrum with those of 2 (Fig. S1.2). Both compounds displayed similar Cotton effect, which suggested that compound 3 had the same (3R) absolute configuration as 2. Thus, the structure of 3 was defined as shown in Fig. 1.
The molecular formula of compound 4 was determined to be C12H14O6 by the HR-ESI–MS ion at m/z 277.0687 [M + Na]+ (calcd for C12H14O6Na+, 277.0683) together with its 1H and 13C NMR data. Its NMR data (Table 2) were similar to those of penicanesin E (8) [15], except that 4 had an additional ethoxy group [δC 15.4 (CH3) and 60.7 (CH2); δH 1.07 (3H, t, J = 7.0 Hz), 3.22 and 3.06 (each 1H, dq, J = 9.0, 7.0 Hz)]. This was supported by the 1H–1H COSY correlations, and the location of the ethoxy group at C-3 was determined by the HMBC correlation of the protons at 3.22 and 3.06 ppm to C-3. The (3R) absolute configuration of 4 was also determined by the ECD calculation (Fig. 5). Thus, the structure of 4 was determined to be the C-3 ethylation product of 8.
Compound 5 was obtained as colorless needles and its molecular formula of C16H20O5 was determined by the HR-ESI-MS and 1D NMR data. The 1D NMR data of 5 (Table 3) were resembled those of penigriseofulvin E (10) [21], with the only difference being the presence of a 1,3,4,5-tetrasubstituted aromatic ring [δH 5.96 and 6.16 (each 1H, d, J = 1.9 Hz)] in 5 instead of a pentasubstituted aromatic ring in 10. The MS data and the key HMBC correlations of H-7 to C-3a and C-5 indicated that 4 was the dechlorination product of 10. The NOE correlation of H-5′/H-1′ suggested that these two protons were cofacial and arbitrarily designated as α-configuration. The relative configuration of C-2 could be the same as 10 by comparison of their 1D NMR data. A single-crystal X-ray diffraction analysis using Cu Kα radiation [Flack parameter = 0.09 (4)] (Fig. 4) further confirmed the structure of 5 including the (2S, 1'S, 6'R) absolute configuration.
The 1H NMR (400 MHz) and 13C NMR (100 MHz) data of 5 and 6 in CDCl3
Compound 6 had a molecular formula C12H14O6 determined by its HR-ESI-MS and 1D NMR data. The 1H NMR data of 6 (Table 3) showed two singlet methyls including a methoxy [δH 2.48 and 3.95 (each 3H, s)], one ethoxy group [δH 1.37 (3H, t, J = 7.1 Hz), and 4.34 (2H, q, J = 7.1 Hz)], and an aromatic proton [δH 6.91 (1H, s)]. The 12 carbon signals in the 13C NMR and DEPT spectra were assigned by 2D NMR data analysis as a pentasubstituted aromatic ring, an acetyl group, an ester carbonyl group, one methoxy group, and ethoxy group. The HMBC correlations from H-6 to C-4, C-2, and C-7, H3-9 to C-2 and C-8, 5-OMe to C-5, and H-1′ to C-7 from H-2 to C-7 confirmed that the acetyl group at C-1, the ester carbonyl with an ethoxy group at C-1, the methoxy group at C-5, and two hydroxy groups at C-3 and C-4 of the aromatic ring (Fig. 3). Thus, the structure of 6 was determined as shown.
The planar structure of compound 7 was determined to be the same as 4,5,6-trihydroxy-3-methylphthalide, a racemic mixture ([α]D25 = 0) [20], by comparison of their 1D NMR data. The experimental ECD curve of 7 was completely consistent with compounds 2 and 3, so as their optical rotations (7: [α]D25 = +34.8, 2: [α]D25 = +36.9, 3: [α]D25 = +12.5), which suggested that the absolute configuration of 7 was also 3R. Thus, compound 7 was defined as (R)-4,5,6-trihydroxy-3-methylphthalide and named as penicanesol G. In addition, the same planar structure with negative optical rotation [[α]D25 = -25.9] synthesized by Zhang et al. was (S)-4,5,6-trihydroxy-3-methylphthalide [22].
Twenty-seven known compounds were one phthalide derivative, penicanesin E (8) [15]; one phthalan derivative, curvulol (9) [19]; six griseofulvin analogues, penigriseofulvin E (10) [21], dechlorogriseofulvin (11) [23], griseofulvin (12) [24], 4′-demethoxyisogriseofulvin (13) [24], isogriseofulvin (14) [24], and dehydrogriseofulvin (15) [24]; five 2-acetyl-phenylacetic acid derivatives, curvulin acid (16) [25], O-methylcurvulinic acid (17) [26], methyl curvulinate (18) [27], methyl 2-(2-acetyl-3-hydroxy-5-methoxyphenyl)acetate (19) [28], and methyl 2-acetyl-3-hydroxy-4,5-dimethoxybenzeneacetate (20) [29]; three butyrolactone derivatives, butyrolactone Ⅱ (21) [30], butyrolactone Ⅰ (22) [30], and butyrolactone-Ⅴ (23) [31]; two sesterterpenoids, terretonin (24) [32] and terretonin B (25) [33]; three decaturin alkaloids, decaturin D (26) [34], decaturin E (27) [35], and 15-deoxyoxalicine B (28) [36]; four indole alkaloids, tryptoquivaline (29) [37], quinadoline A (30) [38], tryptoquivaline L (31) [39], and tryptoquivaline R (32) [40]; and two diketopiperazines, fumitremorgin C (33) [41] and 6-methoxyspirotryprostatin B (34) [42]. Their structures were identified by comparison of their spectroscopic data (Tables S2.1–S2.16 in Supporting Information) with those reported information.
The selected compounds 1–15 and 21–34 isolated from the fungus P. canescens L1 were evaluated their cytotoxic activities towards NCI-H1975. At a concentration of 20 μM, two compounds (9 and 13) had good cytotoxicity, and 13 had significant activity even at a sub-concentration of 20 μM (Fig. 6). Subsequently, these two active compounds were subjected to evaluate their 50% inhibiting concentration (IC50), and compounds 13 and 9 had the IC50 values 4.24 ± 0.13 μM and 18.02 ± 0.48 μM, respectively. Gefitinib was a positive control drug with an IC50 value of 12.99 ± 0.13 μM.
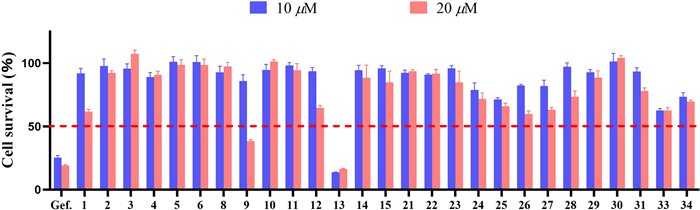
Preliminary screening of the cytotoxic activities of compounds 1–15, 21–31, 33, and 34 against NCI-H1975 cells. Cells were treated with compounds at the initial screening concentration of 10 and 20 μM for 72 h, then cell viability was determined by Cell Counting Kit-8
3 Experimental section
3.1 General experimental procedures
Optical rotations were measured on an Anton Paar MCP200 modular circular polarimeter. UV and ECD spectra were recorded on an Applied Photophysics Chirascan spectrometer. IR spectra were obtained from a PerkinElmer Spectrum Two FTIR spectrometer with a UATR accessory. 1D and 2D NMR spectra were collected from Bruker Ascend TM 500 and Bruker Avance Ⅲ 400 spectrometers at 25 ℃ with TMS as the internal standard. HR-ESI-MS data were acquired via a Waters Micromass Q-TOF spectrometer and a X500R QTOF spectrometer from SCIEX. Semipreparative HPLC was carried out on a Shimadzu LC-20 AT equipped with an SPD-M20A PDA detector. A NanoChrom ChromCoreTM 5–120 C18 column (250 × 10 mm, 5 µm), a Phenomenex Lux cellulose-2 chiral-phase column (250 × 10 mm, 5 μm), and a YMC-pack ODS-A column (250 × 10 mm, S-5 μm, 12 nm) were utilized for HPLC purification. Solvents MeCN for HPLC were purchased from BCR International Trading Co. Ltd. Reversed-phase C18 (Rp-C18) silica gel from YMC Co. Ltd. (12 nm, S-50 μm), MCI gel from Mitsubishi Chemical Industries Ltd. (CHP20P, 75–150 μm), Sephadex LH-20 gel from Amersham Biosciences, and silica gel from Qingdao Haiyang Chemical Co, Ltd. (100–200, 300–400 mesh) were utilized for general column chromatographic separation, and fractions were monitored via silica gel TLC (GF254 plates, 0.25 mm thickness) visualized with 15% sulfuric acid in EtOH.
3.2 Fungal material
The fungal strain L1 was isolated from the fresh roots sample of Croton cnidophyllus and identified as Penicillium canescens on the base of ITS region (GenBank OQ438055). The fungal strain (No. L1) is deposited in School of Pharmaceutical Sciences, Sun Yat-sen University.
3.3 Fermentation and extraction
The strain P. canescens L1 was cultured on potato dextrose agar (PDA) plates (PDB 24.0 g and agar 18.0 g in 1.0 L H2O) at 28 ℃ for 7 days. The seed medium (PDB media 24.0 g in 1.0 L H2O) was inoculated with strain P. canescens L1 and incubated at 28.0 ℃ for 3 days on a rotating shaker (180 rpm). For chemical investigations, a large-scale fermentation of P. canescens L1 was incubated for 32 days at 28 ℃ in 1.5 L × 160 Erlenmeyar flasks (each flask contained 100 g rice and 100 mL H2O). After incubation, every flask was ultrasonically extracted with 4 × 0.4 L 95% EtOH for 30 min. After removing the solvents under vacuum, 250 g of crude extract was obtained, which was then suspended in water (3 L) and successively partitioned with petroleum ether (PE, 2 × 3 L) and EtOAc (3 × 3 L).
3.4 Isolation and purification
The obtained EtOAc fraction (150 g) was firstly separated over a silica gel column eluted with a gradient of PE/EtOAc (50:1 → 1:1) followed by the solvents of DCM/MeOH (70:1 → 0:1) to afford eight fractions (Frs. 1–8).
Fr. 4 (4 g) was separated by Rp-C18 silica gel CC (MeOH/H2O, 20% → 100%) to give five fractions (Frs. 4A–4E). Fr. 4A (1.3 g) was subjected to Sephadex LH-20 CC (MeOH) to yield Fr. 4A1, Fr. 4A2 (150 mg), and compound 9 (87 mg). Compound 3 (22 mg, tR 20 min) was obtained from Fr. 4A2 by semipreparative HPLC equipped with a YMC-pack ODS-A column (MeCN/H2O, 27:73, 3 mL/min). Compound 29 (23 mg) was precipitated from Fr. 4E (160 mg).
Fr. 5 (18.4 g) was subjected to Rp-C18 silica gel CC (MeOH/H2O, 20% → 65%) to give five fractions (Frs. 5A–5E). Fr. 5A was divided into three fractions (Frs. 5A1–5A3) by Sephadex LH-20 CC (MeOH). Fr. 5A2 (4.6 g) was further divided into seven fractions (Frs. 5A2A–5A2G) by silica gel CC (PE/DCM, 1:1 → 0:1 and then DCM/MeOH, 500:1 → 70:1). Fr. 5A2D (661 mg) was separated by Sephadex LH-20 CC (MeOH) to give three fractions (Frs. 5A2D1–5A2D3). Compound 20 (8 mg, tR 10 min) was obtained from Fr. 5A2D1 (17 mg) by semipreparative HPLC equipped with a chiral column (MeCN/H2O, 45:55, 3 mL/min), 19 (58 mg, tR 8 min) was obtained from Fr. 5A2D2 (219 mg) by semipreparative chiral HPLC (MeCN/H2O, 40:60, 3 mL/min), and 18 (19 mg, tR 8 min) was obtained from Fr. 5A2D3 (270 mg) by semipreparative chiral HPLC (MeCN/H2O, 35:65, 3 mL/min). Fr. 5B (824 mg) was separated by silica gel CC (PE/DCM, 1:1 → 0:1 and then DCM/MeOH, 500:1 → 70:1) to give Frs. 5B1–5B4 and compound 17 (10 mg). Compounds 4 (18 mg) and 2 (47 mg) were obtained from Fr. 5B1 (90 mg) by Sephadex LH-20 CC (MeOH). Fr. 5C (934 mg) was further divided into six fractions (Frs. 5C1–5C6) by silica gel CC (PE/DCM, 1:1 → 0:1 and then DCM/MeOH, 500:1 → 70:1). Fr.5C2 (260 mg) was successively purified by Sephadex LH-20 CC (MeOH) and semipreparative HPLC equipped with a NanoChrom ChromCoreTM 5–120 C18 column (MeCN/H2O, 30:70, 3 mL/min) to yield 6 (44 mg, tR 7.5 min). Fr. 5E (3.9 g) was separated by Sephadex LH-20 CC (MeOH) to give three fractions (Frs. 5E1–5E3). Fr. 5E1 (1.93 g) was further divided into three fractions (Frs. 5E1A–5E1C) by silica gel CC (PE/DCM, 1:1 → 0:1 and then DCM/MeOH, 500:1 → 70:1). Compounds 11 (83 mg, tR 7.5 min) and 13 (24 mg, tR 10.5 min) were obtained from Fr. 5E1A (200 mg) by semipreparative HPLC equipped with a NanoChrom ChromCoreTM 5–120 C18 column (MeCN/H2O, 60:40, 3 mL/min). Fr. 5E1B (520 mg) was subjected to Sephadex LH-20 CC (MeOH) and followed by semipreparative chiral HPLC (MeCN/H2O, 53:47, 3 mL/min) to give 5 (14 mg, tR 10 min) and 10 (64 mg, tR 12.5 min). Fr. 5E1C (818 mg) was subjected to silica gel CC (PE/EtOAc, 70:1 → 0:1) to yield 24 (20 mg) and 1 (2 mg). Fr. 5E2 (1.2 g) was separated by silica gel CC (PE/EtOAc, 70:1 → 1:1) to give compound 22 (220 mg) and Frs. 5E2A–5E2E. Fr. 5E2E (63 mg) was further purified by semipreparative chiral HPLC (MeCN/H2O, 50:50, 3 mL/min) to yield 21 (30 mg, tR 6 min) and 23 (2 mg, tR 8 min).
Fr. 6 (24 g) was subjected to Rp-C18 silica gel CC (MeOH/H2O, 20% → 70%) to give six fractions (Frs. 6A–6F). Fr. 6A (5.2 g) was further separated by silica gel CC (DCM/MeOH, 500:1 → 70:1) to yield Frs. 6A1–6A5, compound 8 (84 mg), and Fr. 6A6 (300 mg). Fr. 6A6 was firstly separated by semipreparative chiral HPLC (MeCN/H2O, 15:85, 3 mL/min) to give two fractions (Frs. 6A7A–6A7B). Then, Fr. 6A7A (91 mg) was purified by HPLC equipped with a NanoChrom ChromCoreTM 5–120 C18 column (MeCN/H2O, 15:85, 3 mL/min) to yield 16 (21 mg, tR 9 min), and compound 7 (8 mg, tR 8 min) was obtained from Fr. 6A7B (40 mg) also by HPLC equipped with a NanoChrom ChromCoreTM 5–120 C18 column (MeCN/H2O, 20:80, 3 mL/min). Fr. 6E (4.7 g) was further separated by silica gel CC (DCM/MeOH, 500:1 → 70:1) to give Frs. 6E1–6E9, 28 (23 mg), and 34 (14 mg). Fr. 6E2 (200 mg) was purified by semipreparative HPLC equipped with a YMC-pack ODS-A column (MeCN/H2O, 45:55, 3 mL/min) to yield 12 (22 mg, tR 20 min), 15 (3 mg, tR 21 min), and 14 (14 mg, tR 23.5 min). Fr. 6E5 (510 mg) was separated by Sephadex LH-20 CC (MeOH) to give 25 (34 mg). Fr. 6E6 (500 mg) was subjected to Sephadex LH-20 CC (MeOH) to yield two fractions (Frs. 6E6A–6E6B). Fr. 6E6B (250 mg) was further purified over semipreparative HPLC equipped with a YMC-pack ODS-A column (MeCN/H2O, 35:65, 3 mL/min) to afford 31 (3 mg, tR 17 min) and 33 (1 mg, tR 20 min). Fr. 6E8 (220 mg) was divided into compound 30 (20 mg) and Fr. 6E8A (150 mg) by Sephadex LH-20 CC (MeOH), and then compound 32 (16 mg, tR 17 min) was obtained from Fr. 6E8A by HPLC equipped with a NanoChrom ChromCoreTM 5–120 C18 column (MeCN/H2O, 40:60, 3 mL/min). Fr. 6F (800 mg) was subjected to silica gel CC (PE/DCM, 1:1 → 0:1 and then DCM/MeOH, 500:1 → 70:1) to give compound 26 (140 mg) and Fr. 6F1 (150 mg). Compound 27 (8 mg, tR 16 min) was obtained from Fr. 6F1 by semipreparative HPLC equipped with a YMC-pack ODS-A column (MeCN/H2O, 68:32, 3 mL/min).
3.5 Spectroscopic data of compounds
3.5.1 Penicanesol A (1)
Colorless needles; m.p. 256–257; [α]D25 + 19.9 (ca. 0.09, MeCN); UV (MeCN) λmax (log ε) 230 (3.50) nm; ECD (ca. 2.6 × 10−4 M, MeCN) λmax (Δε) 203 (+4.50), 218 (−2.06), 240 (+ 0.26) nm; IR (UATR) νmax 3357, 2921, 2852, 1705, 1612, 1572, 1496, 1349, 1130 cm−1; 1H and 13C NMR data see Table 1; HR-ESI-MS m/z 411.1055 [M + Na]+ (calcd for C20H20O8Na+, 411.1050).
3.5.2 Penicanesol B (2)
White amorphous solid; [α]D25 + 36.9 (ca. 0.12, MeCN); UV (MeCN) λmax (log ε) 190 (3.50) nm; ECD (ca. 4.8 × 10−4 M, MeCN) λmax (Δε) 193 (+1.84), 216 (−3.70), 231 (+ 1.04), 247 (−0.35) nm; IR (UATR) νmax 3349, 1720, 1630, 1492, 1447, 1353, 1113 cm−1; 1H and 13C NMR data see Table 2; HR-ESI-MS m/z 233.0422 [M + Na]+ (calcd for C10H10O5Na+, 233.0420).
3.5.3 Penicanesol C (3)
White amorphous solid; [α]D25 + 12.5 (c 0.13, MeCN); UV (MeCN) λmax (log ε) 190 (3.70) nm; ECD (ca. 4.5 × 10−4 M, MeCN) λmax (Δε) 193 (+1.96), 216 (−4.63), 231 (+ 1.11), 247 (−0.37) nm; IR (UATR) νmax 3353, 1645, 1516, 1489, 1043 cm−1; 1H and 13C NMR data see Table 2; HR-ESI-MS m/z 225.0743 [M + H]+ (calcd for C11H13O5+, 225.0757), 247.0565 [M + Na]+ (calcd for C11H12O5Na+, 247.0577).
3.5.4 Penicanesol D (4)
White amorphous solid; [α]D25 + 44.9 (c 0.10, MeCN); UV (MeCN) λmax (log ε) 210 (4.80) nm; ECD (ca. 3.9 × 10−4 M, MeCN) λmax (Δε) 193 (+2.22), 216 (−5.25), 231 (+ 1.25) nm; IR (UATR) νmax 3361, 2921, 2851, 1738, 1629, 1497, 1462, 1184 cm−1; 1H and 13C NMR data see Table 2; HR-ESI-MS m/z 277.0687 [M + Na]+ (calcd for C12H14O6Na+, 277.0683).
3.5.5 Penicanesol E (5)
Colorless needles; [α]D25 + 58.9 (ca. 0.11, MeCN); UV (MeCN) λmax (log ε) 254 (4.70) nm; ECD (ca. 3.4 × 10−4 M, MeCN) λmax (Δε) 192 (−12.06), 215 (+ 15.63), 233 (−3.04), 280 (+ 2.60), 329 (−4.75) nm; IR (UATR) νmax 3479, 1681, 1615, 1596, 1459, 1216, 1155 cm−1; 1H and 13C NMR data see Table 3; HR-ESI-MS m/z 315.1206 [M + Na]+ (calcd for C16H20O5Na+, 312.1203).
3.5.6 Penicanesol F (6)
White amorphous solid; UV (MeCN) λmax (log ε) 254 (4.81) nm; 1H and 13C NMR data see Table 3; HR-ESI-MS m/z 277.0678 [M + Na]+ (calcd for C12H14O6Na+, 277.0683).
3.5.7 Penicanesol G (7)
White amorphous solid; [α]D25 + 34.8 (ca. 0.14, MeCN); UV (MeCN) λmax (log ε) 216 (3.49), 238 (2.50), 266 (2.98) nm; ECD (ca. 5.1 × 10−4 M, MeCN) λmax (Δε) 193 (+1.71), 216 (−4.05), 231 (+ 0.97), 247 (−0.32) nm; IR (UATR) νmax 3275, 1702, 1621, 1522, 1497, 1317, 1043 cm−1; 1H and 13C NMR data see Table S2.1 in Supporting Information; HR-ESI-MS m/z 197.0441 [M + H]+ (calcd for C12H15O6+, 197.0444).
3.6 Crystallographic data
Compounds 1, 5, 8, and 13 were recrystallized MeOH/DCM (5:1) to afford colorless needles at room temperature. Crystallographic data for the structures determined in this study have been deposited at the Cambridge Crystallographic Data Centre (deposition number: 2402573 for 1, 2402743 for 5, 2407293 for 8, and 2402745 for 13) and can be obtained free of charge from the CCDC Web site (https://www.ccdc.cam.ac.uk/).
Penicanesol A (1):Crystal Data for C20H20O8 (M = 388.36 g/mol):orthorhombic, space group P212121 (no. 19), a = 8.79670(9) Å, b = 12.20777(15) Å, c = 16.4867(2) Å, V = 1770.47(4) Å3, Z = 4, T = 100.00(10) K, μ(Cu Kα) = 0.959 mm−1, Dcalc = 1.457 g/cm3, 18, 304 reflections measured (9.014° ≤ 2θ ≤ 158.02°), 3748 unique (Rint = 0.0477, Rsigma = 0.0310) which were used in all calculations. The final R1 was 0.0372 (I > 2σ(I)) and wR2 was 0.1020 (all data). Flack parameter = 0.02(8).
Penicanesol E (5):Crystal Data for C16H20O5 (M = 292.32 g/mol):monoclinic, space group P21 (no. 4), a = 6.08950(10) Å, b = 10.3226(2) Å, c = 11.9172(2) Å, β = 104.211(2)°, V = 726.18(2) Å3, Z = 2, T = 99.99(10) K, μ(Cu Kα) = 0.818 mm−1, Dcalc = 1.337 g/cm3, 7450 reflections measured (7.652° ≤ 2Θ ≤ 156.932°), 2703 unique (Rint = 0.0154, Rsigma = 0.0132) which were used in all calculations. The final R1 was 0.0270 (I > 2σ(I)) and wR2 was 0.0716 (all data). Flack parameter = 0.09 (4).
Penicanesin E (8):Crystal Data for C10H10O6 (M = 226.18 g/mol):monoclinic, space group P21/c (no. 14), a = 8.30853(9) Å, b = 14.86270(18) Å, c = 7.72506(8) Å, β = 97.8579(10)°, V = 944.989(18) Å3, Z = 4, T = 100.00(10) K, μ(Cu Kα) = 1.155 mm−1, Dcalc = 1.590 g/cm3, 18, 746 reflections measured (10.75° ≤ 2Θ ≤ 157.508°), 2007 unique (Rint = 0.0443, Rsigma = 0.0214) which were used in all calculations. The final R1 was 0.0365 (I > 2σ(I)) and wR2 was 0.0983 (all data).
4′-Demethoxyisogriseofulvin (13):Crystal Data for C16H15ClO5 (M = 322.73 g/mol):orthorhombic, space group P212121 (no. 19), a = 11.3451(2) Å, b = 16.9711(3) Å, c = 38.8580(6) Å, V = 7481.7(2) Å3, Z = 20, T = 100.00(10) K, μ(Cu Kα) = 2.462 mm−1, Dcalc = 1.433 g/cm3, 73, 432 reflections measured (4.548° ≤ 2Θ ≤ 157.614°), 15, 741 unique (Rint = 0.0862, Rsigma = 0.0645) which were used in all calculations. The final R1 was 0.0948 (I > 2σ(I)) and wR2 was 0.2488 (all data). Flack parameter = 0.034 (7). Flack parameter = 0.09 (4).
3.7 ECD calculations
Please see S1.1 in Supporting Information for details of the quantum chemical ECD calculation of 2 and 4.
3.8 Cytotoxic activity assay
For cell viability, NCI-H1975 cells were seeded in 96-well plates at 2,000 cells per well (optimum density for growth) in a total volume of 100 μL of media containing 10% serum. Serially diluted compounds in 50 μL of media were added to the cells 24 h later. After 3 days of incubation, Cell Counting Kit-8 reagents (Dojindo, Japan) were added, and luminescence was measured according to the manufacturer's instructions. All experiments were repeated three times. The data are presented as percentage of viable cells with vehicle-treated cells set as 100. The estimated in vitro IC50 values were calculated by using GraphPad Prism 9 software.
4 Conclusion
In summary, the chemical study of P. canescens L1 resulted in the identification of seven novel polyketides labelled as penicanesols A–G (1–7), and penicanesol A (1) was a rare dimer derived from phthalan derivatives among them. In the activity screen, compounds 1–15 and 21–34 showed different level of cytotoxic activities towards NCI-H1975. Compounds 9 and 13 had significant activities at a concentration of 20 μM, and 13 (IC50 = 4.24 ± 0.13 μM) exhibited cytotoxicity superior to that of the positive drug gefitinib (IC50 = 12.99 ± 0.13 μM). These findings not only expand the structural diversity of the polyketides but also deepen our understanding of the chemical and bioactivity diversity of plant-derived fungus P. canescens L1.
Notes
Acknowledgements
This work was supported by the Science and Technology Program of Guangzhou, China (No. 2024B03J1322), the Science and Technology Planning Project of Guangdong Province, China (No. 2023A1111120025), the National Natural Science Foundation of China (Nos. 82404454 and 22407144), and the Guangdong Basic and Applied Basic Research Foundation, China (No. 2021B1515140062).
Author contributions
WYW and XW carried out the experiments; QL and YFF wrote the manuscript; LMW screened the biological activities; LL did the ECD calculation; SQW, FYY, and DH analyzed the results; QRL, ZHS, and TY assisted in analyzing the results; GHT designed and checked the whole manuscript. All authors read and approved the final manuscript.
Availability of data and materials
All data generated and analyzed during this study are included in this published article and its Additional file 1.
Declarations
Competing interests
The authors declare that there are no competing interests associated with this work.
References
-
1.Zhang X, Yin Q, Li X, Liu X, Lei H, Wu B. Structures and bioactivities of secondary metabolites from Penicillium genus since 2010. Fitoterapia. 2022;163: 105349. CrossRef PubMed Google Scholar
-
2.Wang C, Lu H, Lan J, Zaman K, Cao S. A review: halogenated compounds from marine fungi. Molecules. 2021;26(2): 458. CrossRef PubMed Google Scholar
-
3.Sang M, Feng P, Chi L-P, Zhang W. The biosynthetic logic and enzymatic machinery of approved fungi-derived pharmaceuticals and agricultural biopesticides. Nat Prod Rep. 2024;41(4): 565-603. CrossRef PubMed Google Scholar
-
4.Lv F, Zeng Y. Novel bioactive natural products from marine-derived Penicillium fungi: a review (2021–2023). Mar Drugs. 2024;22(5): 191. CrossRef PubMed Google Scholar
-
5.Agrawal S, Chavan P, Badiger A. Marine fungi of the genera Aspergillus and Penicillium: a promising reservoir of chemical diversity for developing anti-viral drug candidates. The Microbe. 2024;3: 100081. CrossRef PubMed Google Scholar
-
6.Zang Y, Gong Y, Gong J, Liu J, Chen C, Gu L, Zhou Y, Wang J, Zhu H, Zhang Y. Fungal polyketides with three distinctive ring skeletons from the fungus Penicillium canescens uncovered by OSMAC and molecular networking strategies. J Org Chem. 2020;85(7): 4973-80. CrossRef PubMed Google Scholar
-
7.Yaegashi J, Romsdahl J, Chiang YM, Wang CCC. Genome mining and molecular characterization of the biosynthetic gene cluster of a diterpenic meroterpenoid, 15-deoxyoxalicine B, in Penicillium canescens. Chem Sci. 2015;6(11): 6537-44. CrossRef PubMed Google Scholar
-
8.Wang JP, Shu Y, Zhang SQ, Yao LL, Li BX, Zhu L, Zhang X, Xiao H, Cai L, Ding ZT. Polyketides with antimicrobial activities from Penicillium canescens DJJ-1. Phytochemistry. 2023;206: 113554. CrossRef PubMed Google Scholar
-
9.Nicoletti R, Lopez-Gresa MP, Manzo E, Carella A, Ciavatta ML. Production and fungitoxic activity of Sch 642305, a secondary metabolite of Penicillium canescens. Mycopathologia. 2007;163(5): 295-301. CrossRef PubMed Google Scholar
-
10.Malik A, Ardalani H, Anam S, McNair LM, Kromphardt KJK, Frandsen RJN, Franzyk H, Staerk D, Kongstad KT. Antidiabetic xanthones with α-glucosidase inhibitory activities from an endophytic Penicillium canescens. Fitoterapia. 2020;142: 104522. CrossRef PubMed Google Scholar
-
11.Frank M, Hartmann R, Plenker M, Mandi A, Kurtan T, Oezkaya FC, Mueller WEG, Kassack MU, Hamacher A, Lin W, Liu Z, Proksch P. Brominated azaphilones from the sponge-associated fungus Penicillium canescens strain 4.14.6a. J Nat Prod. 2019;82(8): 2159-66. CrossRef PubMed Google Scholar
-
12.Dasanayaka SAHK, Nong XH, Liang X, Liang JQ, Amin M, Qi SH. New dibenzodioxocinone and pyran-3,5-dione derivatives from the deep-sea-derived fungus Penicillium canescens SCSIO z053. J Asian Nat Prod Res. 2020;22(4): 338-45. CrossRef PubMed Google Scholar
-
13.Bertinetti BV, Pena NI, Cabrera GM. An antifungal tetrapeptide from the culture of Penicillium canescens. Chem Biodivers. 2009;6(8): 1178-84. CrossRef PubMed Google Scholar
-
14.Zang Y, Gong YH, Li XW, Li XN, Liu JJ, Chen CM, Zhou Y, Gu LH, Luo ZW, Wang JP, Sun WG, Zhu HC, Zhang YH. Canescones A-E: aromatic polyketide dimers with PTP1B inhibitory activity from Penicillium canescens. Org Chem Front. 2019;6(18): 3274-81. CrossRef PubMed Google Scholar
-
15.Zang Y, Gong Y, Shi Z, Qi C, Chen C, Tong Q, Liu J, Wang J, Zhu H, Zhang Y. Multioxidized aromatic polyketides produced by a soil-derived fungus Penicillium canescens. Phytochemistry. 2022;193: 113012. CrossRef PubMed Google Scholar
-
16.Wei X, Huang JL, Gao HH, Yuan FY, Tang GH, Yin S. New halimane and clerodane diterpenoids from Croton cnidophyllus. Nat Prod Bioprospect. 2023;13(1): 21. CrossRef PubMed Google Scholar
-
17.Chen JQ, Li S, Fan RZ, Sun ZH, Zhu XY, Yin AP, Tang GH, Yin S. Talaesthanes A–C, three new meroterpenoids from the endophytic fungus Talaromyces primulinus H21. Fitoterapia. 2024;177: 106085. CrossRef PubMed Google Scholar
-
18.Wang Y, Song SH, Wu LM, Zhou X, Lu QR, Yin AP, Yin S, Tang GH. Chemical constituents of Penicillium ferraniaense GE-7 and their cytotoxicities. Nat Prod Res. 2024. CrossRef PubMed Google Scholar
-
19.Liu T, Li ZL, Wang Y, Zhang LM, Song JL, Tian L, Pei YH, Hua HM. Study on the secondary metabolites of marine-derived fungus Penicillium sacculum. Chin Pharm J. 2012;47(8): 577-80. PubMed Google Scholar
-
20.Mullady EL, Millett WP, Yoo HD, Weiskopf AS, Chen J, DiTullio D, Knight-Connoni V, Hughes DE, Pierceall WE. A phthalide with in vitro growth inhibitory activity from an Oidiodendron strain. J Nat Prod. 2004;67(12): 2086-9. CrossRef PubMed Google Scholar
-
21.Liang Y, Zhang B, Li D, Chen X, Wang Q, Shu B, Li Q, Tong Q, Chen C, Zhu H, Zhang Y. Griseofulvin analogues from the fungus Penicillium griseofulvum and their anti-inflammatory activity. Bioorg Chem. 2023;139: 106736. CrossRef PubMed Google Scholar
-
22.Chen W, Jiang J, Wang J. Asymmetric ruthenium-catalyzed C−H activation by a versatile chiral-amide-directing strategy. Angew Chem Int Ed. 2024;63(6): e202316741. CrossRef PubMed Google Scholar
-
23.Zhao JH, Zhang YL, Wang LW, Wang JY, Zhang CL. Bioactive secondary metabolites from Nigrospora sp. LLGLM003, an endophytic fungus of the medicinal plant Moringa oleifera Lam. World J Microbiol Biotechnol. 2012;28(5): 2107-12. PubMed Google Scholar
-
24.Levine SG, Hicks RE, Gottlieb HE, Wenkert E. Carbon-13 nuclear magnetic resonance spectroscopy of naturally occurring substances. XXX. Griseofulvin J Org Chem. 1975;40(17): 2540-2. CrossRef PubMed Google Scholar
-
25.Wen X, Zhang DW, Guo SX, Wang CL. Chemical constituents of an endophytic fungus Chaetosphaeronema sp. from Phlomis younghusbandii Mukerjee. Chin Med Biotechnol. 2014;9(6): 453-6. PubMed Google Scholar
-
26.Ying YM, Zhang LW, Shan WG, Zhan ZJ. Secondary metabolites of Peyronellaea sp. XW-12, an endophytic fungus of Huperzia serrata. Chem Nat Comp. 2014;50(4): 723-5. PubMed Google Scholar
-
27.Ichinose K, Ebizuka Y, Sankawa U. Mechanistic studies on the biomimetic reduction of tetrahydroxynaphthalene, a key intermediate in melanin biosynthesis. Chem Pharm Bull. 2001;49(2): 192-6. CrossRef PubMed Google Scholar
-
28.Beekman AM, Barrow RA. Stereochemical assignment of the fungal metabolites pestalotiopsones D and E through enantiopure synthesis. J Nat Prod. 2013;76(11): 2054-9. CrossRef PubMed Google Scholar
-
29.Kamal A, Qureshi AA, Ahmad A, Rickards RW. Biochemistry of microorganisms. V. Structure and synthesis of curvulic acid and curvin, metabolic products of Curvularia siddiqui. Tetrahedron. 1965;21(6): 1411. CrossRef PubMed Google Scholar
-
30.Gong T, Dong SH, Zhu P. Butyrolactone derivatives isolated from the marine fungus Aspergillus versicolor F62. Mycosystema. 2014;33(3): 706-12. PubMed Google Scholar
-
31.Nagia MMS, El-Metwally MM, Shaaban M, El-Zalabani SM, Hanna AG. Four butyrolactones and diverse bioactive secondary metabolites from terrestrial Aspergillus flavipes MM2: isolation and structure determination. Org Med Chem Lett. 2012;2(1): 9. CrossRef PubMed Google Scholar
-
32.Matsuda Y, Iwabuchi T, Wakimoto T, Awakawa T, Abe I. Uncovering the unusual D-ring construction in terretonin biosynthesis by collaboration of a multifunctional cytochrome P450 and a unique isomerase. J Am Chem Soc. 2015;137(9): 3393-401. CrossRef PubMed Google Scholar
-
33.Li GY, Li BG, Yang T, Yin JH, Qi HY, Liu GY, Zhang GL. Sesterterpenoids, terretonins A−D, and an alkaloid, asterrelenin, from Aspergillus terreus. J Nat Prod. 2005;68(8): 1243-6. CrossRef PubMed Google Scholar
-
34.Li C, Gloer JB, Wicklow DT, Dowd PF. Antiinsectan decaturin and oxalicine analogues from Penicillium thiersii. J Nat Prod. 2005;68(3): 319-22. CrossRef PubMed Google Scholar
-
35.Wang PL, Li DY, Xie LR, Wu X, Hua HM, Li ZL. Novel decaturin alkaloids from the marine-derived fungus Penicillium oxalicum. Nat Prod Commun. 2013. CrossRef PubMed Google Scholar
-
36.Zhang Y, Li C, Swenson DC, Gloer JB, Wicklow DT, Dowd PF. Novel antiinsectan oxalicine alkaloids from two undescribed fungicolous Penicillium spp. Org Lett. 2003;5(5): 773-6. CrossRef PubMed Google Scholar
-
37.Liu H, Yan C, She Z. Study on the isolation, identification and anti-inflammatory activity of the secondary metabolites of alkaloids produced by S apetala-derived fungi Aspergillus sp ZJ-S4. J Guangdong Pharma Univ. 2021;37(3): 1-6. PubMed Google Scholar
-
38.Koyama N, Inoue Y, Sekine M, Hayakawa Y, Homma H, Omura S, Tomoda H. Relative and absolute stereochemistry of quinadoline B, an inhibitor of lipid droplet synthesis in macrophages. Org Lett. 2008;10(22): 5273-6. CrossRef PubMed Google Scholar
-
39.Buttachon S, Chandrapatya A, Manoch L, Silva A, Gales L, Bruyère C, Kiss R, Kijjoa A. Sartorymensin, a new indole alkaloid, and new analogues of tryptoquivaline and fiscalins produced by Neosartorya siamensis (KUFC 6349). Tetrahedron. 2012;68(15): 3253-62. CrossRef PubMed Google Scholar
-
40.Xu N, Cao Y, Wang L, Chen G, Pei YH. New alkaloids from a marine-derived fungus Neosartorya sp. HN-M-3. J Asian Nat Prod Res. 2013;15(7): 731-6. CrossRef PubMed Google Scholar
-
41.Cui CB, Kakeya H, Osada H. Novel mammalian cell cycle inhibitors, tryprostatins A, B and other diketopiperazines produced by Aspergillus fumigatus. Ⅱ. Physico-chemical properties and structures. J Antibiot. 1996;49(6): 534-40. CrossRef PubMed Google Scholar
-
42.Zhang M, Wang WL, Fang YC, Zhu TJ, Gu QQ, Zhu WM. Cytotoxic alkaloids and antibiotic nordammarane triterpenoids from the marine-derived fungus Aspergillus sydowi. J Nat Prod. 2008;71(6): 985-9. CrossRef PubMed Google Scholar
Copyright information
© The Author(s) 2025
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
