


Emestrin-type epipolythiodioxopiperazines from Aspergillus nidulans with cytotoxic activities by regulating PI3K/AKT and mitochondrial apoptotic pathways
National Natural Science Foundation of China (U22A20380, 82173706, 82104028) and Fundamental Research Funds for the Central Universities (2024BRA018) financially supported this project
Abstract
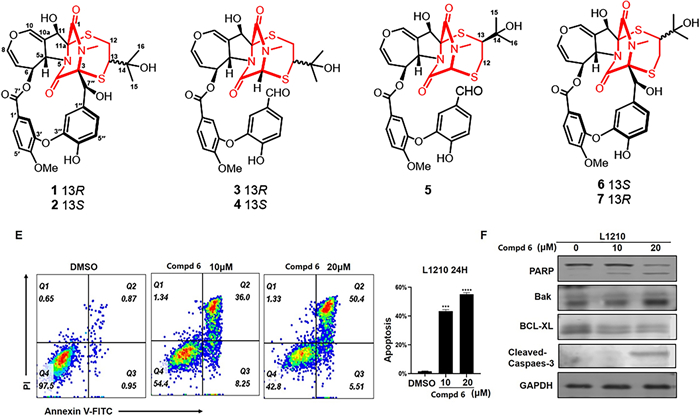
Five novel emestrin-type epipolythiodioxopiperazines (ETPs), prenylemestrins C−G (1–5), along with two known ETPs, prenylemestrin A (6) and prenylemestrin B (7), were obtained from Aspergillus nidulans. Their structures were characterized by spectroscopic data, X-ray crystallographic data, ECD comparisons and calculations. Prenylemestrins C−G (1 − 5) represent a rare class of ETPs, characterized by a 2,5-dithia-7,9-diazabicyclo[4.2.2]decane-8,10-dione core involving a hemiterpene moiety. Notably, compound 6 exhibited moderate cytotoxicity, inducing G2/M cell cycle arrest and apoptosis of L1210 cells by regulating the PI3K/AKT signaling pathway and mitochondrial apoptotic mechanisms.Graphical Abstract
Keywords
Aspergillus nidulans Epipolythiodioxopiperazines Thioethanothio bridge Structural elucidation Cytotoxicity1 Introduction
Fungal natural products are a crucial source for drug discovery, serving as a rich reservoir for novel bioactive compounds [1–3]. Their fascinating structural diversity and biological activities hold out the ideal choices for the development of novel agents, and they also help discover novel structures, providing effective agents for the treatment of various human diseases [2, 4–6]. Epipolythiodioxopiperazines (ETPs) represent a distinct class of fungal metabolites, characterized by a di- or polysulfide-bridged dioxopiperazine ring, and derived from two amino acids. These compounds are renowned for broad-spectrum of bioactivities, including cytotoxicity against tumor cells, antibacterial function, antiviral, immunologic suppression, and anti-inflammatory action [7–10]. Based on the structure of the core ETP moiety and the modifications, emestrins are a notable group of monomeric ETPs, distinguished by a dihydrooxepino[4,3-b]pyrrole core and 15-membered macrolide [11]. Emestrin, first obtained from Emericella striata in 1985, demonstrated antifungal activity and cytotoxicity via DNA fragmentation in HL-60 cells [12–15]. To date, numerous emestrin-type ETPs have been identified, with their structural uniqueness and promising bioactivities drawing increasing scientific interest [16–22]. For instance, secoemestrin C exhibits potent anti-cancer activity in GEM-resistant and GEM-sensitive pancreatic adenocarcinoma (PAAD) cells, inducing mitochondria-mediated apoptosis and causing severe endoplasmic reticulum damage [23, 24].
Previously, we identified four emestrin hybrid polymers, asperemestrins A–D, and a highly productive ETP, secoemestrin C, which exhibited notable immunosuppressive effect, from Aspergillus nidulans [19, 25]. Recently, we found that the first cysteine-retained emestrin, nidustrin A, features a unique sulfur-containing 18-membered macrocyclic lactone [26]. Building on this work, our ongoing efforts to explore bioactive ETPs have led to the discovery of prenylemestrins C−G (1 − 5). These compounds are distinguished by a 2,5-dithia-7,9-diazabicyclo[4.2.2]decane-8,10-dione core involving a hemiterpene moiety (Fig. 1). In this study, the isolation, structural elucidation, and biological activities of these compounds were described in detail.

Chemical structures of 1−7
2 Results and discussion
Prenylemestrin C (1) was isolated as a white powder with the molecular formula C32H32N2O11S2, based on the HRESIMS spectrum with an ion peak at m/z 707.1345 ([M + Na]+, calcd. for 707.1345), indicating 18 degrees of unsaturation. The IR spectrum revealed the presence of hydroxyl (3419 cm−1), carbonyl (1691 and 1662 cm−1), and aromatic ring (1513 cm−1) functionalities. The 1H NMR data of 1 recorded in CD3OD (Table 1) showed signals corresponding to two 1,3,4-trisubstituted benzene rings protons at δH 7.98 (d, J = 2.0 Hz), 7.81 (dd, J = 8.6, 2.0 Hz), and 7.19 (d, J = 8.6 Hz), and δH 8.42 (d, J = 2.3 Hz), 7.28 (dd, J = 8.6, 2.3 Hz), and 6.90 (d, J = 8.6 Hz), seven methines including three olefinic [δH 6.92 (d, J = 2.4 Hz), 6.42 (dd, J = 8.1, 2.4 Hz), and 5.01 (dd, J = 8.1, 2.3 Hz)] and three oxymethines [δH 5.49 (dt, J = 8.3, 2.3 Hz), 5.12 (s), and 4.72 (s)], one methylene [δH 3.46 (dd, J = 16.5, 1.8 Hz) and 2.89 (dd, J = 16.5, 5.3 Hz)], a methoxy group (δH 3.99), an N-methyl (δH 3.36), and two singlet methyls (δH 0.80, 0.68). The 13C NMR and DEPT spectroscopic data of 1 recorded in CD3OD displayed 32 carbon signals (Table 2), including three carbonyls (δC 167.2, 167.1, 161.5), 10 nonprotonated carbons (seven olefinic and three oxygenated sp3 at δC 78.0, 76.5, 73.8), 14 methines (nine olefinic and five sp3), a methylene (δC 35.3), and four methyls (δC 56.8, 29.0, 26.1, 28.4).
1H NMR Spectroscopic Data (400 MHz; δ in ppm, J in Hz) for Compounds 1–5
13C NMR Spectroscopic Data (100 MHz; δ in ppm) for Compounds 1–5
The planar structure of compound 1 was elucidated through 2D NMR spectral analysis. The independent spin system of H-5a/H-6/H-7/H-8, along with HMBC correlations of H-10/C-5a, C-8, and C-10a, confirmed the presence of a 6,7-dihydrooxepine ring. Further HMBC spectrum showed correlations from H-11 to C-5a, C-10a, C-11a, and C-1, as well as from H-5a to C-4 and from the N-methyl protons to C-1 and C-3. These findings established that the 6,7-dihydrooxepine ring was fused to a dioxopiperazine moiety via an 11-hydroxypyrrolidine ring. Additionally, a 3′-oxygen-4′-methoxy-benzoate fragment and a 3′′-oxygen-4′′-hydroxybenzyl unit were identified as being connected to the 6,7-dihydrooxepine ring at C-6 and the dioxopiperazine ring at C-3, respectively. This connectivity was supported by the HMBC correlations from H-6 to C-7′ and from H-7′′ to C-3, C-4, C-1′′, C-2′′, and C-6′′. Moreover, the 1H−1H COSY correlation of H2-12/H-13, combined with the HMBC cross-peak from H2-12 to C-11a, H-13 to C-3, H3-15/H3-16 to C-13 and C-14, confirmed the attachment of an isopentan-3-ol fragment to C-11a and C-3 through sulfur atoms. Consequently, compound 1 was deduced to feature an epipolythiodioxopiperazine skeleton with a thioethanothio bridge, closely resembling the planar structure with prenylemestrins A and B (6 and 7) [17]. A detailed comparison of their 1H and 13C NMR data revealed that the key difference lies in the 2,5-dithia-7,9-diazabicyclo[4.2.2]decane-8,10-dione core. HMBC correlations (Fig. 2) from H2-12 to C-11a and from H-13 to C-3 confirmed that C-12 and C-13 were linked to C-11a and C-3, respectively. With the hemiterpene moiety switched, 1 can also be interpreted as a migration of the 2-hydroxyisopropyl group.
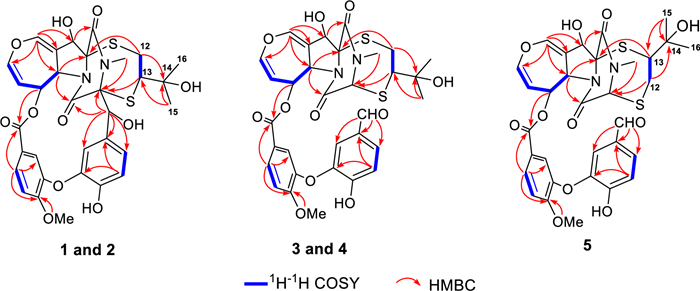
Key 1H−1H COSY and HMBC correlations of compounds 1−5
To determine the relative configuration of compound 1, NMR data were recorded in DMSO-d6 (Tables 1 and 2). The trans-orientation of H-5a and H-6 was confirmed by the large coupling constant (J = 8.3 Hz) observed between those protons [21]. The NOESY correlation between H-5a and OH-11 indicated that H-11 and H-6 were on the same side, tentatively assigned as α orientation [17]. Furthermore, NOESY correlations (Fig. 3) of H-6/H-13, H-2′/Me-15/Me-16, H-2″/Me-15/Me-16, suggested that the configuration of C-13 should be R*. Accordingly, the C-3 − S and C-11a − S bonds were deduced to be α-oriented. Additionally, NOESY interactions of N-Me/H-7″, H-7″/H-2″, and H-7″/H-6″, OH-7″/H-6″, and OH-7″/N-Me, supported the assignment of the configurations at C-7′′ as S*. Based on these observations, the relative configuration of 1 was established.
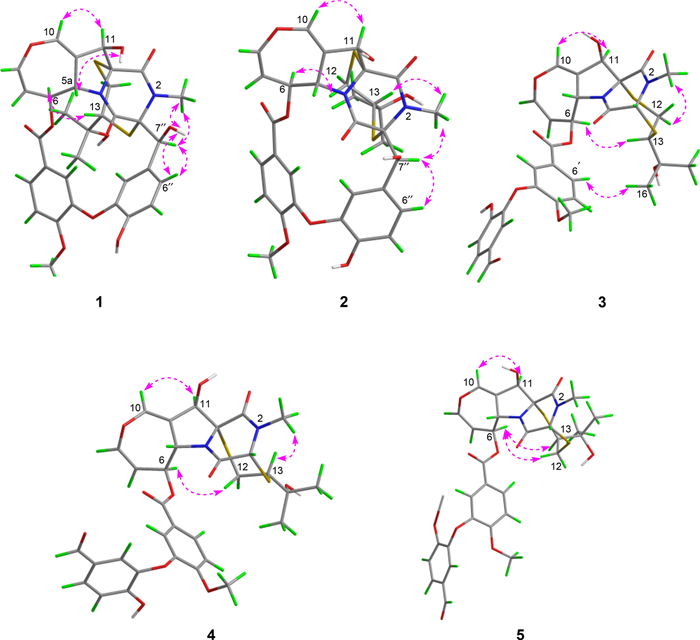
Key NOESY correlations of compounds 1−5
Prenylemestrin D (2) was determined to share the same molecular formula, C32H32N2O11S2, as compound 1, based on HRESIMS and NMR spectra. A comparison of the 1D (Tables 1 and 2) and 2D NMR spectra (Fig. 3) revealed significant similarities between the two compounds, except for the slightly different chemical shifts of C-1 (2: δC 170.4; 1: δC 167.1), C-4 (2: δC 165.4; 1: δC 161.5), and C-11a (2: δC 75.3; 1: δC 78.0), which inferred that 2 could be the C-13 epimer of 1 (Fig. 1). This deduction was further supported by NOESY interactions of H-13/N-Me and H-6/H-12a (δH 2.76) in 2. Additionally, the experimental ECD spectra (Fig. 4) of compounds 1 and 2 closely matched those of 6 and 7, whose absolute configurations had been previously confirmed by X-ray diffraction (Fig. 5). These findings conclusively established the absolute configurations of 1 and 2 as depicted in Fig. 1.
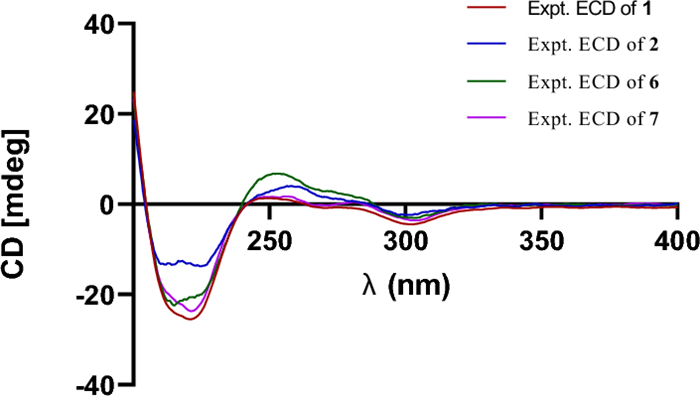
Experimental ECD spectra of compounds 1, 2, 6, and 7
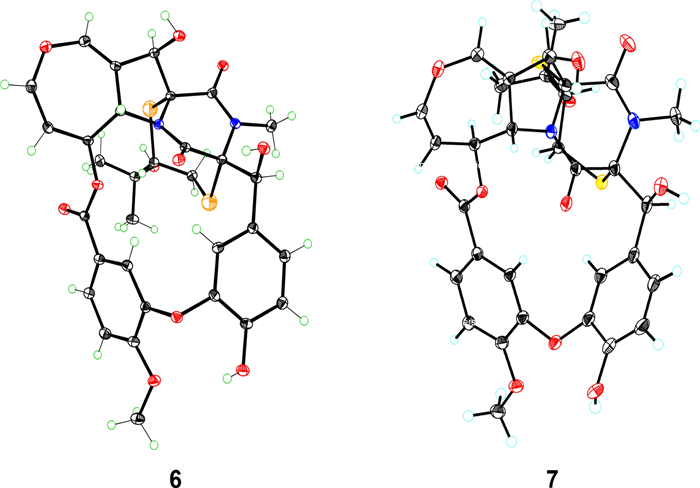
ORTEP drawing of the X-ray crystal structures of 6 and 7
Prenylemestrin E (3) was assigned the molecular formula of C32H32N2O11S2, as indicated by its HRESIMS spectrum, showing an ion peak at m/z 707.1372 ([M + Na]+, calcd. for C32H32N2O11S2Na+, 707.1345), corresponding 18 degrees of unsaturation. The 1D NMR data of 3 closely resembled those of 1 and 2 (Tables 1 and 2), except for the appearance of distinctive signals for a conjugated aldehyde group (δC 190.9 and δH 9.75), and a methine (C-3, δH 4.96, s; δC 65.7), which replaced the nonprotonated carbon C-3 (δC 76.5) in 1. These differences suggested that 3 was structurally related to secoemestrin C, with cleavage of the 15-membered macrolactone ring between C-3 and C-7′′. HMBC correlations from H-7′′ to C-1′′, C-2′′, and C-6′′ confirmed the presence and position of an aldehyde group. Moreover, HMBC interactions from H-13 to C-3, and from H2-12 to C-11a along with the 1H − 1H COSY correlations of H2-12/H-13 further established the caged core structure of 3 as consistent with that of 1 and 2. The relative configuration of 3 was deduced from the NOESY spectrum and coupling constant analysis. The coupling constants (JH‑5a/H‑6 = 8.0 Hz) indicated that H-5a was trans-oriented. The NOESY correlation of between H-11 and H-6 indicated that H-11 is α-oriented. The observed NOESY correlation (Fig. 3) of H-6/H-13 suggested that the configuration of C-13 was R*, with the C-3−S and C-11a−S bonds being α-oriented, and H-3 being β-oriented. Furthermore, the theoretical 13C NMR calculations with DP4 + analysis of two isomers13R*-3 and 13S*-3 were conducted through the GIAO method at the mPW1PW91/6-311G(d, p) level in chloroform with the Gaussian 09 software. The results (Fig. S1) showed that 13R*-3 had a better coefficient of determination (R2 = 0.9983) between the experimental and calculated 13C NMR chemical shits than 13S*-3 (R2 = 0.9964). Additionally, the DP4 + analysis with a high probability of 100% permitted the relative configuration 13R*-3. To further confirm the absolute configuration of 3, the ECD spectra of (3R*,5aS*,6S*,11R*,11aR*,13R*)-3 were calculated using time-dependent density functional theory (TDDFT) methods (Tables S3 and S4). The calculated spectra matched well with the experimental data (Fig. 6), leading to the assignment of the absolute configurations of 3 as 3R,5aS,6S,11R,11aR,13R.
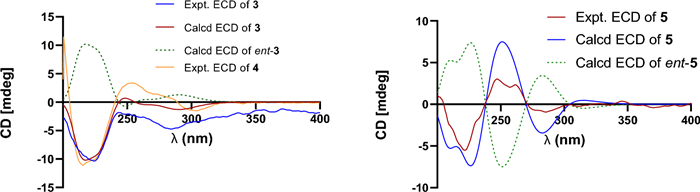
Calculated and experimental ECD spectra of 3 and 5
Prenylemestrin F (4) was determined to have the same molecular formula, C32H32N2O11S2, as compound 3, as confirmed by HRESIMS and NMR data. The 1H and 13C NMR data (Tables 1 and 2) of 4 closely resembled those of 3, indicating a similar structural framework. Further interpretation of the 1H − 1H COSY and HMBC spectra (Fig. 2) confirmed that compound 4 shares the same ETPs core as 3. Notable differences between 3 and 4 were observed in the chemical shifts of C-1 and C-4, which were significantly downfield-shifted by + 4.5 ppm and + 4.9 ppm, respectively, in 3. Detailed 1H − 1H COSY and HMBC analyses confirmed that both compounds possess the same planar structure. Therefore, 3 and 4 were concluded to be stereoisomers. The NOESY interaction of H-13/N-Me was observed for 4, as opposed to H-13/H-6 in 3, which, along with careful NOESY elucidation, suggested that 4 is the C-13 epimer of 3. Furthermore, the experimental ECD spectra (Fig. 6) of compound 4 closely matched those of 3, leading to the determination of the absolute configuration of 4 as 3R,5aS,6S,11R,11aR,13S.
Prenylemestrin G (5) was assigned the same molecular formula of C32H32N2O11S2 as 3 and 4, based on HRESIMS analysis. The 1H and 13C NMR data of 5 (Tables 1 and 2) closely resembled those of 3. HMBC correlations (Fig. 2) from H2-12 to C-3, H-13 to C-11a confirmed that the connection between the hemiterpene moiety and the ETP core in 5 was consistent with prenylemestrins A and B (6 and 7) [17]. NOESY correlations of H-6/H-13 (Fig. 3) indicated that H-13 in 5 adopts an α-orientation. The absolute configuration of 5 was determined by comparison its experimental ECD spectrum with the calculated one. The calculated ECD spectrum for (3R,5aS,6S,11R,11aR,13S)-5 was in good agreement with the experimental data (Fig. 6), leading to the assignment of the absolute configuration as 5 as 3R,5aS,6S,11R,11aR,13S.
Based on previous biosynthetic studies of ETPs, hypothetic biogenetic pathways of 1−7 were proposed to explain their origins (Scheme S1) with two molecules of L-phenylalanine as precursors. The key intermediate Ⅰ was obtained via a series of peptide cyclization, ring-expansion, and esterification reactions. Then, intermediate Ⅰ underwent methylation and macrocyclization to form Ⅱ. Sulfurization of the intermediate Ⅱ at C-3 and C-11a produced the key dithiol intermediate Ⅲ, which was further decorated with dimethylallyl diphosphate via pathway a or b to form Ⅲ or Ⅴ, respectively. Subsequently, the followed epoxidation and nucleophilic attack of the sulfhydryl group led to the production of 1−2 and 6−7. The intermediate Ⅵ, with the C-3−C-7′′ bond cleavaged, could be derived from Ⅲ; ultimately, compounds 3−5 could be derived via pathways c and d, with dimethylallyl diphosphate and modified farnesyl diphosphate, respectively.
In our biological evaluation, compounds 1−7 exhibited no anti-inflammatory activity. Additionally, compounds 1−5 and 7 showed no cytotoxicity against the tested cell lines (A549, L1210, HL-60, SW-480, and Hep3B). Interestingly, 6 demonstrated moderate cytotoxicity against L1210 mouse leukemia cells. Using L1210 cells as a model, we further investigated the antitumor effect and the underlying mechanism of compound 6. The results revealed that 6 inhibited proliferation and induced G2/M cell cycle arrest in L1210 cells by regulating PI3K/AKT pathway and cell cycle-related proteins, such as p-Chk1, Cyclin B, Cdc2, p-Cdc2, and γh2ax (Fig. 7). Furthermore, 6 inhibited mitochondrial membrane potential (MMP), increased ROS levels, and induced apoptosis in L1210 cells (Fig. 8). These changes in MMP and ROS levels suggest that the antitumor effects of 6 are closely related to mitochondrial dysfunction. Western blot analysis further supported this hypothesis, showing that 6 altered the expression of mitochondria-related proteins, including Bak and Bcl-xl, indicating that apoptosis induced by 6 occurs predominantly via the mitochondrial pathway. The antitumor effects of compound 6 were also validated in other leukemia cell lines, with results consistent with those observed in L1210 cells (data not shown). These findings highlight the potential of 6 as a candidate for anti-leukemia research, warranting further in-depth studies on its mechanism and therapeutic potential.
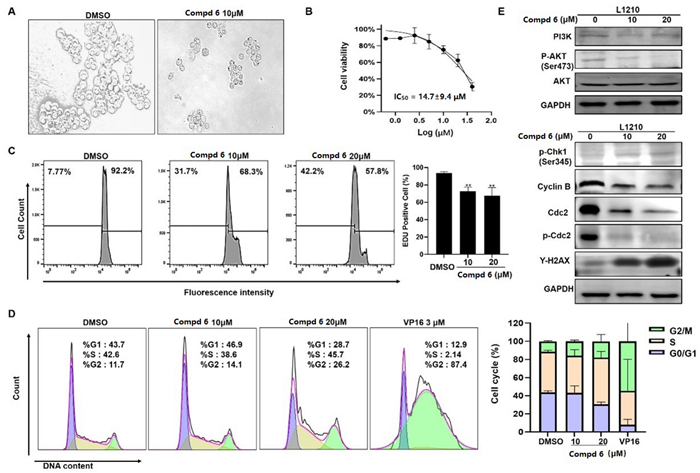
Compound 6 inhibited proliferation and induced G2/M cell cycle arrest in L1210 cells. Morphologic change of L1210 cells after treatment with 6 (A); Cell proliferation curve of CCK-8 viability assay (B); EdU assay showed that EdU positive ratio was affected by 6 treated (C). Cell cycle distribution (D); Western blot analysis of PI3K/AKT and cell cycle related proteins, with GAPDH was used as loading control (E)
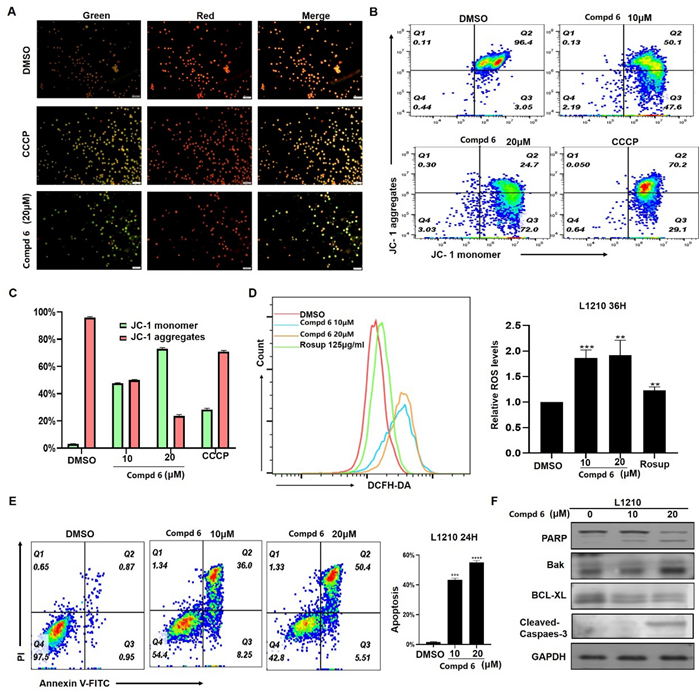
Effect of compound 6 on L1210 cells. Treat L1210 cells with different concentrations of 6 for 24 h and detect mitochondrial membrane potential status using JC-1 staining kit (A−C). D Fluorescence data of DCFH-DA staining of cells treated with 6 or Rosup for 36 h. E Cell apoptosis was determined by flow cytometric analysis using Annexin Ⅴ-FITC and PI staining. F Western blot analysis of apoptosis related proteins, with GAPDH used as loading control
3 Conclusion
This study elucidates the structures of prenylemestrins C−G (1−5) and highlights the moderate cytotoxicity of compound 6, which is likely attributed to its distinct structural features. Compound 6 was found to induce G2/M cell cycle arrest and apoptosis in L1210 cells, primarily through the regulation of the signaling pathway and mitochondrial apoptotic mechanisms. These findings underscore its significant impact on cell growth and survival. The discovery of prenylemestrins C−G expands the structural diversity of epipolythiodioxopiperazine (ETP) compounds and provides a solid foundation for investigating their pharmacological and medicinal potential. Future research should focus on a detailed exploration of the mechanism of action of compound 6 and its potential therapeutic applications in cancer treatment. Additionally, the biological activities and pharmacological properties of other prenylemestrins, as well as their interactions with cellular signaling pathways, warrant further study.
In conclusion, this work not only uncovers the structures and bioactivities of novel ETP compounds but also paves the way for their exploration in synthetic and pharmacological research, fostering interest in their potential applications in drug discovery and development.
4 Experimental
4.1 General experimental procedures
Using Shanghai SGW® X-4B microscopic melting point instrument (uncorrected, China) and hot stage melting point determination to measure melting points. HRESIMS was performed on a microOTOF-Q-Ⅱ acquisition parameter (Bruker, Germany). Measure the optical rotations of compounds in methanol or dichloromethane using a Perkin Elmer 341 polarimeter. The ultraviolet (UV) spectrum was obtained with the Lambda 35 UV-1 spectrometer (PerkinElmer, US). CD curve was obtained on the J-810 spectrometer (JASCO, Japan). IR spectrum was acquired on a Nicolet iS50R FT-IR spectrometer (Thermo Scientific, US). NMR spectrum was recorded on an AM-400 and an AVANCE NEO 600 spectrometer (Bruker, Germany), with chemical shifts are expressed in ppm relative to CD3OD (δC 49.0, δH 3.31) and CDCl3 (δC 77.16, δH 7.26). Silica gel (100–200 mesh and 200–300 mesh, Qingdao Marine Chemical Inc., Qingdao, China) was used for normal-phase column chromatography (CC). Reversed-phase CC was accomplished using YMC-Gel (50 μm, YMC, Japan), and Sephadex LH-20 (Pharmacia Biotech AB, Uppsala, Sweden) were used for CC performed with methanol/dichloromethane as the mobile phase. Thin-layer chromatography (TLC) analyses were carried out with precoated silica gel F254 plates (Qingdao Marine Chemical Inc., Qingdao, China). The HPLC separations were conducted on an Agilent 1220 HPLC system equipped with a UV detector, accompanied by reversed-phase Ultimate XB-C18 column (5 μm, 10 × 250 mm). 10% H2SO4 in EtOH was used to visualize spots in TLC.
4.2 Fungal material
The fungus Aspergillus nidulans, deposited at the Huazhong University of Science and Techbology (HUST, 414JZ-1), Hubei Province, China, was isolated from the Annelida Whitmania pigra Whitman. The ITS sequence data for this strain have been submitted to DDBJ/EMBL/GenBank under accession No. MK397763.
4.3 Fermentation, extraction, and isolation
The fungus A. nidulans was maintained as seeds on potato dextrose agar (PDA) medium at 28 ℃ for 6 days, which was carried out on rice medium at room temperature for three weeks. After incubation, extract the fermented rice substrate seven times with ethanol. After removing the solvent under vacuum, the entire extract was suspended in water and then extracted with ethyl acetate to obtain a dark solid (400.0 g). Based on the TLC analysis, dark solid was chromatographed over silica gel using petroleum ether/ethyl acetate (20:1, 10:1, 5:1, 3:1, 1:1, 0:1, v/v) and ethyl acetate/methanol gradient (60:1 to methanol, v/v) as eluting solvent, to yield six subfractions (Fr.1−Fr.6). Fr.3 (1.5 g) was further separated through Sephadex LH-20 by eluting with dichloromethane/methanol (1:1, v/v), to produce three major fractions (Fr.3.1–Fr.3.3), Fr.3.3 (210 mg) was further separated through a silica gel column eluted with dichloromethane/methanol (100:1, 80:1, 50:1, v/v) and semipreparative HPLC to yield 4 (acetonitrile/water, 65:35, 2.0 mL/min; tR 14.0 min, 2 mg). The Fr.4 (400 mg) was fractionated on a silica gel column eluted with dichloromethane/methanol (200:1, 100:1, 80:1, 50:1, v/v) to yield six major fractions (Fr.4.1–Fr.4.6), Fr.4.2 (100 mg) was further separated via a Sephadex LH-20 CC in methanol and purified by HPLC to obtain compound 6 (acetonitrile/water, 46:54, 2.0 mL/min; 27 mg; tR 39.3 min), Fr.4.3 (89.2 mg) was separated by a Sephadex LH-20 CC in dichloromethane/methanol (1:1, v/v) and further purified by HPLC to yield compounds 2 (acetonitrile/water, 40:60, 2.0 mL/min; 10.2 mg; tR 35.2 min) and 5 (acetonitrile/water, 42:58, 2.0 mL/min; 11.5 mg; tR 13.0 min). The Fr.5 (12 g) was subjected to an reverse phase C18 silica gel eluted with a gradient methanol/water (20%, 40%, 60%, 80%, 100%, v/v) to yield eight fractions (Fr.5.1−Fr.5.8), Fr.5.3 (260 mg) was separated through Sephadex LH-20 eluted with dichloromethane/methanol (1:1, v/v), and followed by semipreparative HPLC to yield compounds 1 (acetonitrile/water, 40:60, 2.0 mL/min; tR 36.0 min, 20 mg) and 3 (acetonitrile/water, 40:60, 2.0 mL/min; tR 48.5 min, 1.9 mg). Fr.5.6 (2.3 g) was chromatographed on a silica gel column by eluting with dichloromethane/methanol (100:1, 80:1, 60:1, v/v) to afford three fractions Fr.5.6.1 − Fr.5.6.3. Fr.5.6.2 (146.0 mg) was further purified to semipreparative HPLC to yield 7 (acetonitrile/water, 30:70, 2.0 mL/min; tR 36.0 min, 38.8 mg).
Prenylemestrin C (1): White powder, [α]D20 –99 (c 0.1, MeOH); UV (MeOH) λmax (log ε) = 203 (4.90), 263 (4.33) nm; IR (KBr) νmax = 3419, 2942, 2838, 1691, 1662, 1513, and 1381 cm−1; ECD (MeOH) λmax (Δε) = 221 (–50.7), 275 (–1.4), 302 (–8.8) nm; 1H and 13C NMR data see Tables S1 and S2; HRMS (ESI-TOF) m/z: [M + Na]+ 707.1345 (calcd for C32H32N2O11S2Na, 707.1345).
Prenylemestrin D (2): White powder; [α]D20 –29 (c 0.1, MeOH); UV (MeOH) λmax (log ε) = 262 (4.08), 202 (4.62) nm; IR (KBr) νmax = 3423, 2974, 2954, 2934, 1675, 1653, 1513, and 1383 cm−1; ECD (MeOH) λmax (Δε) = 225 (–34.3), 258 (+ 10.0), 303 (–5.9) nm; 1H and 13C NMR data see Tables S1 and S2; HRMS (ESI-TOF) m/z: [M + Na]+ 707.1377 (calcd for C32H32N2O11S2Na, 707.1345).
Prenylemestrin E (3): White powder; [α]D20 –15 (c 0.1, MeOH); UV (MeOH) λmax (log ε) = 264 (4.33), 228 (4.50), 203 (4.68) nm; IR (KBr) νmax = 3430, 2920, 2850, 1681, 1606, 1512, and 1384 cm−1; ECD (MeOH) λmax (Δε) = 224 (–25.8), 284 (–11.8) nm; 1H and 13C NMR data see Tables S1 and S2; HRMS (ESI-TOF) m/z: [M + Na]+ 707. 1372 (calcd for C32H32N2O11S2Na, 707.1345).
Prenylemestrin F (4): White powder; [α]D20 –67 (c 0.1, MeOH); UV (MeOH) λmax (log ε) = 263 (4.38), 226 (4.56) nm; IR (KBr) νmax = 3429, 2922, 2850, 1681, 1605, 1511, and 1277 cm−1; ECD (MeOH) λmax (Δε) = 215 (–55.8), 253 (+ 16.8), 304 (–7.6) nm; 1H and 13C NMR data see Tables S1 and S2; HRMS (ESI-TOF) m/z: [M + Na]+ 707.1433 (calcd for C32H32N2O11S2Na, 707.1345).
Prenylemestrin G (5): White powder; [α]D20 +51 (c 0.1, MeOH); UV (MeOH) λmax (log ε) = 263 (4.43), 204 (4.92) nm; IR (KBr) νmax = 3363, 2920, 2850, 1683, 1662, 1604, 1511, and 1277 cm−1; ECD (MeOH) λmax (Δε) = 221 (–59.0), 256 (+ 4.27), 274 (–0.2), 281 (+ 1.2) nm; 1H and 13C NMR data see Tables S1 and S2; HRMS (ESI-TOF) m/z: [M + Na]+ 707.1338 (calcd for C32H32N2O11S2Na, 707.1345).
4.4 X-ray crystal structure analysis
After crystallization from methanol using the vapor diffusion method, colorless crystals of 6 and 7 were obtained at room temperature. Collect crystal data using Bruker D8 Quest (Bruker, Germany) with a graphite-monochromated Cu Kα radiation. The structure was solved with the SHELXTL refinement packages using least squares minimization. All crystallographic data were stored in the Cambridge Crystallographic Data Centre (2133579 for 6 and 2133560 for 7).
Crystal data of 6. C32H32N2O11S2, M = 684.72, space group P212121, a = 9.416(0) Å, b = 10.339(0), c = 33.754(0), α = β = γ = 90°, V = 3286.0(3) Å3, T = 293(2) K, Z = 4, μ(Cu Kα) = 2.012 mm−1, 83,642 reflections measured, 6631 independent reflections (Rint = 0.0325). The final R1 values = 0.0332 (I > 2σ(I)), wR(F2) values = 0.1116 (I > 2σ(I)), R1 values = 0.0334 (all data), and wR(F2) values = 0.1118 (all data). The goodness of fit on F2 = 1.044. Flack parameter = 0.023(4). m.p. 181 − 184 ℃.
Crystal data of 7. C32H32N2O11S2•4(H2O), M = 748.71, space group P43212, a = 18.8206(4) Å, b = 18.8206(4) Å, c = 20.6776(4) Å, α = β = γ = 90°, V = 7324.3(3) Å3, T = 100(2) K, Z = 8, μ(Cu Kα) = 1.938 mm−1, 108,024 reflections measured, 7232 independent reflections (Rint = 0.0743). The final R1 values = 0.0807 (I > 2σ(I)), wR(F2) values = 0.2240 (I > 2σ(I)), R1 values = 0.1109 (all data), and wR(F2) values = 0.2508 (all data). The goodness of fit on F2 = 1.562. Flack parameter = 0.021(7). m.p. 188 − 190 ℃.
4.5 Computational section for electronic circular dichroism (ECD)
The methodology utilized for the theoretical computation of ECD spectra, as well as the production of corresponding calculated ECD spectra, remained consistent with the previously outlined procedure[19]. The details were described in the supplementary material.
Notes
Acknowledgements
The Analytical and Testing Center and Medical Subcenter at HUST is acknowledged for the NMR data and the ECD, UV, and IR spectra collection. We thank the HPC Platform of HUST for assistance in completing computation.
Author contributions
P.L. and Q.L. isolated and purified compounds, identified them, and wrote the original manuscript; A.F. provided assistance in processes such as extraction, isolation, and structural characterisation. Y.X. performed a portion of the pharmacological experiments. C.C. and H.Z. were responsible for the verification and optimization of the manuscript and the delivery of the manuscript. W.W. and Y.Z. guided the experiments. Y-H.Z. supervised, acquired the funding, and revised the manuscript. All authors above reviewed this manuscript.
Data availability
The datasets used or analysed during the current study are available from the corresponding author on reasonable request.
Declarations
Competing interests
The authors declare no competing financial interests.
References
-
1.Abdel-Razek AS, El-Naggar ME, Allam A, Morsy OM, Othman SI. Microbial natural products in drug discovery. Processes. 2020;8(4): 470. CrossRef PubMed Google Scholar
-
2.Newman DJ, Cragg GM. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J Nat Prod. 2020;83(3): 770-803. CrossRef PubMed Google Scholar
-
3.Rodrigues T, Reker D, Schneider P, Schneider G. Counting on natural products for drug design. Nat Chem. 2016;8(6): 531-41. CrossRef PubMed Google Scholar
-
4.Uzma F, Mohan CD, Hashem A, Konappa NM, Rangappa S, Kamath PV, Singh BP, Mudili V, Gupta VK, Siddaiah CN, Chowdappa S, Alqarawi AA, Abd_Allah EF. Endophytic fungi-alternative sources of cytotoxic compounds: a review. Front Pharmacol. 2018;9: 309/301-309/337. PubMed Google Scholar
-
5.Yao H, Liu J, Xu S, Zhu Z, Xu J. The structural modification of natural products for novel drug discovery. Expert Opin Drug Discov. 2017;12(2): 121-40. CrossRef PubMed Google Scholar
-
6.Li Q, Zheng Y, Fu A, Wei M, Kang X, Chen C, Zhu H, Zhang Y. 30-norlanostane triterpenoids and steroid derivatives from the endophytic fungus Aspergillus nidulans. Phytochemistry. 2022;201: 113257. CrossRef PubMed Google Scholar
-
7.Huber EM. Epipolythiodioxopiperazine-based natural products: building blocks, biosynthesis and biological activities. ChemBioChem. 2022;23(23): e202200341. CrossRef PubMed Google Scholar
-
8.Wang L, Jiang Q, Chen S, Wang S, Lu J, Gao X, Zhang D, Jin X. Natural epidithiodiketopiperazine alkaloids as potential anticancer agents: recent mechanisms of action, structural modification, and synthetic strategies. Bioorg Chem. 2023;137: 106642. CrossRef PubMed Google Scholar
-
9.Martínez C, García-Domínguez P, Álvarez R, de Lera AR. Bispyrrolidinoindoline epi(poly)thiodioxopiperazines (BPI-ETPs) and simplified mimetics: structural characterization, bioactivities, and total synthesis. Molecules. 2022;27(21): 7585. CrossRef PubMed Google Scholar
-
10.Zhu M, Zhang X, Huang X, Wang H, Anjum K, Gu Q, Zhu T, Zhang G, Li D. Irregularly bridged epipolythiodioxopiperazines and related analogues: sources, structures, and biological activities. J Nat Prod. 2020;83(6): 2045-53. CrossRef PubMed Google Scholar
-
11.Onodera H, Hasegawa A, Tsumagari N, Nakai R, Ogawa T, Kanda Y. MPC1001 and its analogues: new antitumor agents from the fungus Cladorrhinum species. Org Lett. 2004;6(22): 4101-4. CrossRef PubMed Google Scholar
-
12.Ueno Y, Umemori K, Niimi E-C, Tanuma S-I, Nagata S, Sugamata M, Ihara T, Sekijima M, Kawai K-I, et al. Induction of apoptosis by T-2 toxin and other natural toxins in HL-60 human promyelotic leukemia cells. Nat Toxins. 1995;3(3): 129-37. CrossRef PubMed Google Scholar
-
13.Seya H, Nakajima S, Kawai K, Udagawa S. Structure and absolute configuration of emestrin, a new macrocyclic epidithiodioxopiperazine from Emericella striata. J Chem Soc, Chem Commun. 1985;10: 657-8. CrossRef PubMed Google Scholar
-
14.Kawai K, Ishizaki K, Nakamaru T, Hisada K, Nozawa Y, Kawai KI. Toxicity of emestrin, a new macrocyclic dithiodioxopiperazine mycotoxin, to mitochondrial function. Mycotoxin Res. 1989;5(1): 2-8. CrossRef PubMed Google Scholar
-
15.Kawahara N, Nozawa K, Nakajima S, Kawai K-I. Aurantioemestrin from Emericella striata and silvathione from Aspergillus silvaticus, possible key intermediates from epidithiodioxopiperazines to trioxopiperazines. J Chem Soc Chem Commun. 1986;19: 1495-6. CrossRef PubMed Google Scholar
-
16.Herath HMTB, Jacob M, Wilson AD, Abbas HK, Nanayakkara NPD. New secondary metabolites from bioactive extracts of the fungus Armillaria tabescens. Nat Prod Res. 2013;27(17): 1562-8. CrossRef PubMed Google Scholar
-
17.Chang S, Cai M, Xiao T, Chen Y, Zhao W, Yu L, Shao R, Jiang W, Zhang T, Gan M, Si S, Chen M. Prenylemestrins A and B: Two unexpected epipolythiodioxopiperazines with a thioethanothio bridge from Emericella sp. isolated by genomic analysis. Org Lett. 2022;24(32): 5941-5. CrossRef PubMed Google Scholar
-
18.Chen Y, Xiao T, Guo S, Chang S, Xi X, Su B, Zhang T, Yu L, Zhao W, Wu J, Li Y, Si S, Chen M. Unexpected noremestrin with a sulfur-bearing 15-membered macrocyclic lactone from Emericella sp. 1454. Org Lett. 2024;26(1): 1-5. CrossRef PubMed Google Scholar
-
19.Li Q, Fu A, Wei M, Xiao Y, Yin J, Huang J, Li X-N, Tong Q, Chen C, Zhu H, Zhang Y. Asperemestrins A-D, emestrin hybrid polymers with bridged skeletons from the endophytic fungus Aspergillus nidulans. Org Lett. 2022;24(37): 6800-4. CrossRef PubMed Google Scholar
-
20.Li Y, Yue Q, Krausert NM, An Z, Gloer JB, Bills GF. Emestrins: Anti-cryptococcus epipolythiodioxopiperazines from Podospora australis. J Nat Prod. 2016;79(9): 2357-63. CrossRef PubMed Google Scholar
-
21.Lv F-Y, Mándi A, Li X-M, Chi L-P, Li X, Wang B-G, Kurtán T, Meng L-H. Emestrin-type thiodiketopiperazines from Aspergillus nidulans SD-531, a fungus obtained from the deep-sea sediment of cold seep in the South China Sea. Deep Sea Res, Part Ⅰ. 2023;195: 104004. CrossRef PubMed Google Scholar
-
22.Wu J-S, Shi X-H, Yao G-S, Shao C-L, Fu X-M, Zhang X-L, Guan H-S, Wang C-Y. New thiodiketopiperazine and 3,4-dihydroisocoumarin derivatives from the marine-derived fungus Aspergillus terreus. Mar Drugs. 2020;18(3): 132. CrossRef PubMed Google Scholar
-
23.Ooike M, Nozawa K, Kawai K-I. An epitetrathiodioxopiperazine related to emestrin from Emericella foveolata. Phytochemistry. 1997;46(1): 123-6. CrossRef PubMed Google Scholar
-
24.Wang J, Chen M, Wang M, Zhao W, Zhang C, Liu X, Cai M, Qiu Y, Zhang T, Zhou H, Zhao W, Si S, Shao R. The novel ER stress inducer Sec C triggers apoptosis by sulfating ER cysteine residues and degrading YAP via ER stress in pancreatic cancer cells. Acta Pharm Sin B. 2022;12(1): 210-27. CrossRef PubMed Google Scholar
-
25.Tan X, Sun L, Li Q, Qi C, Fu C, Zhu H, Yang X, Feng H, Li Y, Zhang Y, Chen G. Secoemestrin C inhibits activation of NKT/conventional T cells and protects against concanavalin A-induced autoimmune hepatitis in mice. Am J Transl Res. 2020;12(7): 3389-401. PubMed Google Scholar
-
26.Fu A, Li Q, Li Y, Chen Y, Wei Y, Dong J, Peng Y, Deng M, Sun W, Chen C, Zhang Y, Zhu H. Nidustrin A cysteine-retained emestrin with a unique 18-membered macrocyclic lactone from the endophytic fungus Aspergillus nidulans. Bioorg Chem. 2025;155: 108105. CrossRef PubMed Google Scholar
Copyright information
© The Author(s) 2025.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
