


Design, synthesis and biological evaluation of buthutin derivatives as cardioprotective agents

Abstract
Natural products are the important sources in cardiovascular drug development. In this study, twenty-nine buthutin derivatives were designed, synthesized, and evaluated for their NHE-1 inhibition and protective effects on cardiomyocyte injury. The structure of the newly synthesized compounds had been confirmed by 1H-NMR, 13C-NMR, and HR-ESI-MS spectra. Among all target compounds at 1 μM, compounds 9d, 9f, 9k, 9m, and 9n, with a protection ratio exceeding 30%, exerted stronger protective effects on H9c2 cardiomyocyte than positive control dexrazoxane and buthutin A. Meanwhile, compounds 9k, 9m, and 9o showed the significant NHE-1 inhibitory activities on H9c2 cardiomyocyte, all with a dpHi/min value less than 0.23. What is more, compounds 9k, 9m, 9o and buthutin A all exhibited the specificity on NHE-1 inhibition. Molecular modelling studies suggested the ability of compounds 9m and 9o to establish interactions with three hydrogen bonds to Asp267 and Glu346 of NHE-1, but also the ability with much lower CDOCKER energies than positive control cariporide and buthutin A. The structure–activity relationship (SAR) studies suggested that the presences of amide group, four-carbon linker, and para hydroxyl benzene ring were advantageous pharmacophores for above two pharmacological actions. This research would open new avenues for developing amide-guanidine-based cardioprotective agents.Graphical Abstract
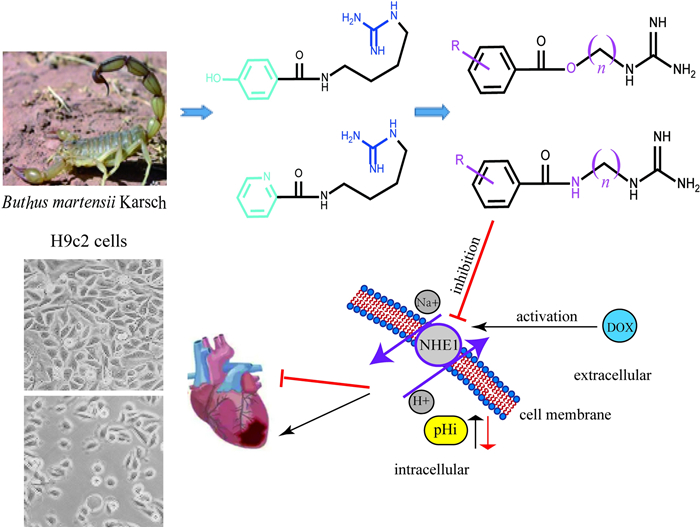
Keywords
Buthus martensii Amide-guanidine derivatives Cardioprotective agents NHE-11 Introduction
The Na+ /H+ exchangers (NHEs) are a group of membrane proteins that transport one Na+ into the cells and one H+ out of the cells in order to regulate the intracellular pH and cell volume [1, 2]. There are about ten NHEs isoforms, and NHE-1 is the major isoform expressed in the heart, which plays a key role in the progression of cardiac hypertrophy to heart failure [3]. It was reported that the increased NHE-1 gene expression led to the increased NHE-1 activity, which ultimately was participated in the cardiac hypertrophy [4]. Besides, the over-expression of NHE-1 in H9c2 cardiomyocytes could also induce myocardial hypertrophy [5]. In contrast, the myocardial NHE-1 expression knockdown by the specific siRNA could significantly alleviate the myocardial hypertrophy of rats [6].
As known, doxorubicin (Dox) is a broad-spectrum anthracycline drug widely used in the treatment of several solid tumors (such as breast cancer, ovarian cancer and gastrointestinal malignancies) and hematological malignancies (such as lymphoma and leukemia) in adults and children [7]. However, Dox can also lead to severe cardiotoxicity, including irreversible degenerative cardiomyopathy and heart failure [8]. Some researches reported that Dox could cause cardiotoxicity because of triggering free radical release, calcium overload, increased NHE-1 activity, apoptosis and so on [9-11]. At present, dexrazoxane (Dex) was the only drug approved by Food and Drug Administration (FDA) to alleviate the cardiotoxicity caused by Dox [12]. Notably, selective inhibition of NHE-1 could also alleviate the Dox-induced cardiotoxicity in rats [13]. Considering Dex’s adverse effects such as the secondary malignancies induction [14], it is worthy of further developing innovative drugs for Dox-induced cardiotoxicity considering the role of NHE-1 inhibition.
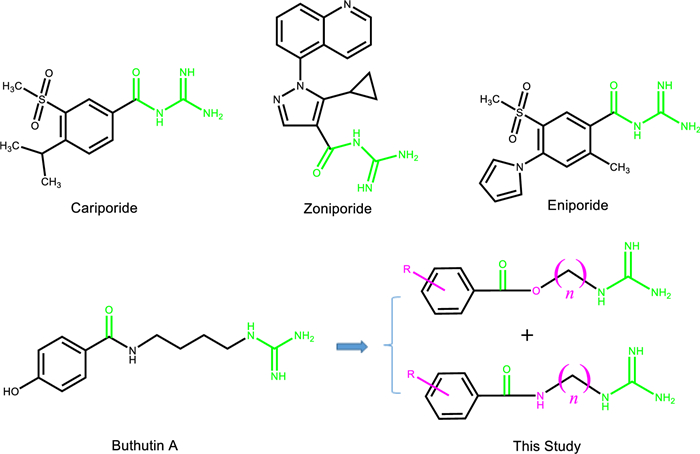
Now the structural types of selective NHE-1 inhibitors are mainly acylguanidine, and the animal experiments have shown that they have a good protective effect on myocardial ischemia and reperfusion injury, and can reduce the mortality of myocardial infarction, infarction size and arrhythmia occurrence [15]. It was reported that cariporide, eniporide and zoniporide (Fig. 1) had entered the clinical stage, but the clinical investigations of cariporide and eniporide didn’t obtain the expected results [16]. Then it is very meaningful to search for the NHE-1 inhibitors with novel structural types.
Reference NHE-1 inhibitors and design of target compounds
Natural products are the important sources in cardiovascular drug development [17-20]. In the process of our successively developing cardioprotective candidates [21, 22], we found that buthutin A (Fig. 1), previously isolated from Buthus martensii by our team [23], had the protective effect on cardiomyocyte injury caused by Dox and the inhibitory activity on NHE-1 (Table 1). In view of its rare amide-guanidine skeleton, we modified the chemical structure of buthutin A and synthesized twenty-nine compounds. The structural types of above amide-guanidine derivatives were obviously different from that of acylguanidine references, because we embedded different lengths of carbon chains and amino functional groups (or ether bonds) in the middle by keeping apart the acyl group from the guanidine group (Fig. 1). We further examined the inhibitory effects of all synthesized compounds on the NHE-1 activity and protective effects on the H9c2 cardiomyocyte injury induced by Dox. The results showed that some compounds revealed significant protective effects on the H9c2 cardiomyocyte injury induced by Dox, and that they also exerted significant inhibition on NHE-1, which would open new avenues for developing amide-guanidine-based cardioprotective agents.
The effects of all compounds at 1 μM against H9c2 cardiomyocyte injury and NHE-1 activity
2 Results and discussion
2.1 Design and synthesis of buthutin derivatives
Inspired by the evident inhibition of buthutin A on NHE-1, we conducted the molecular docking of buthutin A with NHE-1 by comparison with cariporide. Although being similar binding mode to cariporide, buthutin A with three hydrogen bonds to Asp267 and Glu346, had more binding sites than cariporide (with two hydrogen bonds to Asp267 and Glu346), and caused much lower CDOCKER energy (−34.71 kcal/mol) with NHE-1 in comparison to that (−24.17 kcal/mol) of cariporide (Fig. 2). Cariporide is the best studied specific and selective NHE-1 inhibitor [24]. Interactions of the guanidine group of cariporide with Asp267 are believed to stabilize the complex in the outward-facing state, while Glu346 is a crucial residue involved in interactions with cariporide [24]. Now it was evidenced that the hot-spot NHE-1 residues Asp267 and Glu346 coordinate the Na+ ion in the outward-facing state during ion exchange, because the guanidine group of cariporide is of a similar size and charge with a (partially) hydrated Na+ ion [25]. Accordingly, these modeling studies provided crucial insights for guiding further drug design using buthutin A as lead compound. As shown in Fig. 1, buthutin A can be divided into four regions, i.e., guanidine group, phenyl ring, amide linkage and four-carbon linker, respectively. The guanidine group was kept unchanged, while modifications were focused on the other three parts (Fig. 1). Firstly, introducing diverse substituent groups to phenyl ring or replacement of phenyl ring with pyridine ring prompted the syntheses of compounds 7a–7c and 9i–9o. Additionally, the lengths of carbon chains were varied to synthesize compounds 9a–9h and 9p–9u. Subsequently, only replacement of amide linkage with ester linkage or concurrently adjusting the carbon linker lengths provided compounds 13a–13e.

Binding models of cariporide, buthutin A, compounds 9m and 9o with NHE-1 (PDB: 7DSX). A 3D binding mode of cariporide with NHE-1; B 2D binding mode of cariporide with NHE-1; C 3D binding mode of buthutin A with NHE-1; D 2D binding mode of buthutin A with NHE-1; E 3D binding mode of compound 9m with NHE-1; F 2D binding mode of compound 9m with NHE-1; G 3D binding mode of compound 9o with NHE-1; H 2D binding mode of compound 9o with NHE-1
Synthesis of the target compounds was outlined in Scheme 1. In this study, methyl isothioureide sulfate was used as the starting material, and its amino group was protected by Boc group to obtain compound 4. Then compound 4 was reacted with 1, 3-propanediamine, 1, 4-butanediamine and 1, 5-pentylenediamine (or 3-amino-1-propanol and 4-amino-1-butanol) under certain conditions to give series intermediates 5 (or 10). The hydroxybenzoic acids 1 were protected with the acetyl group beforehand to give acetoxybenzoic acid intermediates 2. Series compounds 5 were subjected to amidation with pyridinic acid and acetoxybenzoic acid intermediates 2 to give compounds 6a–6c and 8a–8u, respectively. After removing the Boc protecting group of compounds 6a–6c, target compounds 7a–7c were obtained. Two kinds of protection groups were removed from compounds 8a–8u with methanesulfonic acid (MSA) or trifluoroacetic acid (TFA) through one step to obtain target compounds 9a–9u. Series compounds 10 were esterified with the acetoxybenzoic acid intermediates 2 to afford compounds 11a–11e. The acetyl protecting groups of compounds 11a–11e were first removed with NaOH solution to obtain the compounds 12a–12e, and then their Boc protection groups were taken off using ZnBr2 to acquire target compounds 13a–13e.
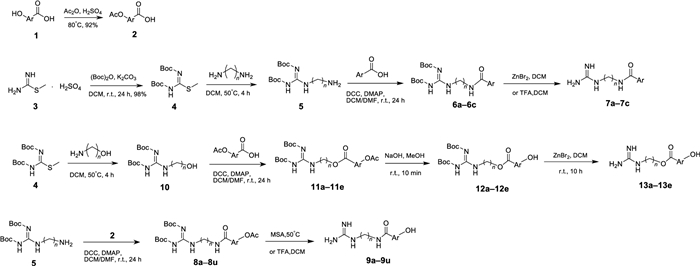
Synthesis of target compounds 7a–7c and 9a–9u and 13a–13e
The structures of all target compounds were confirmed by 1H-NMR, 13C-NMR and HR-ESI–MS spectra. As a representative example, we analyze the spectrum of compound 9h as follows. Its molecular formula was determined to be C11H16N4O4 based on a protonated molecule peak at m/z 269.1272 [M + H]+ (calcd 269.1250 for C11H17N4O4) in its HR-ESI–MS. In its 1H-NMR spectrum, the presence of symmetric benzene ring was confirmed by the single signal with two aromatic protons at δH 6.74. The methylene protons at δH 3.08 and δH 3.23 arose as low field values due to the adjacent nitrogen atoms, and the quintet signal at δH 1.71 showed the presence of a three-carbon aliphatic chain. In its 13C-NMR spectrum, four aromatic carbon resonances at δC 144.6, δC 136.2, δC 124.8, and δC 107.3 indicated one symmetric benzene ring. Characteristic signals at δC 171.2 and δC 158.2 confirmed the presence of an amide group and a guanidine group, respectively. The peaks of three-carbon linker chain were observed at δC 38.7, δC 36.9, and δC 27.6.
2.2 Pharmacological evaluation
In existence of Dox, we examined the cell survival rate of H9c2 cardiomyocyte under the treatment of target compounds and positive control Dex and buthutin A, with the results shown in Table 1. It can be seen that compounds 9d, 9f, 9k, 9m, and 9n at 1 μM showed better protective effects against Dox-induced H9c2 cardiomyocyte injury than Dex and buthutin A, which exerted stronger protective effects with a protection ratio exceeding 30% (Table 1 and Fig. 3).

The effect of compounds 9d, 9f, 9k, 9m, 9n and buthutin A on the morphology of H9c2 cardiomyocyte at the presence of Dox. A Control group; B Dox (1 μM) treated group for 24 h; C Dox (1 μM) and Dex (20 μM) treated group for 24 h; (D) Dox (1 μM) and buthutin A (1 μM) treated group for 24 h; E Dox (1 μM) and 9d (1 μM) treated group for 24 h; F Dox (1 μM) and 9f (1 μM) treated group for 24 h; G Dox (1 μM) and 9 k (1 μM) treated group for 24 h; H Dox (1 μM) and 9m (1 μM) treated group for 24 h; I Dox (1 μM) and 9n (1 μM) treated group for 24 h; J the protection rate of Dex, buthutin A, 9d, 9f, 9k, 9m and 9n on the injury caused by Dox. *p < 0.05, **p < 0.01, and ***p < 0.001 vs Dex group
Taken as an ensemble, the general features of structure activity relationship (SAR) can be deduced from these data in Table 1: (1). By comparing the activities of compounds 7a–7c with the corresponding compounds 9i–9j and buthutin A, it was demonstrated that the aromatic derivatives with electron deficiency were not conducive to the protective effect against myocardial injury. (2). By comparing the activities of compounds 13a–13c with the corresponding compounds 9b–9d, it was shown that the protective effect of amide-guanidine derivatives was stronger than that of ester-guanidine derivatives, which was further verified by the comparison of the activities of compounds 13e and buthutin A. (3). By comparing the activities of compounds 9p–9u with the corresponding compounds 9i–9k and 9n–9o, it was revealed that the derivatives with five-carbon linker were not conducive to the protective effect against myocardial injury. (4). By comparing the activities of compounds 9a–9h with the corresponding compounds 9i–9o, it was illustrated that most derivatives with three-carbon linker possessed a lower protective effect than those with four-carbon linker, except for compound 9f, which had a higher protective effect than 9m. (5). Different positions of hydroxyl groups on the benzene ring in amide-guanidine compounds caused regular variation on protective effects, with the rank order of para- > meta- > ortho-, such as 9c > 9b > 9a or buthutin A > 9j > 9i. (6). In amide-guanidine compounds with a p-hydroxyl group, when another substituent was introduced into its ortho or meta position, respectively, the meta substitution derivatives had a stronger protective effect than those with ortho substitution, such as 9f, 9d > 9e or 9k, 9m > 9l. (7). when the hydroxyl groups of benzene ring were added to the number of three, the bioactivities of amide-guanidine compounds would decrease. For example, the protective effect of 9o was weaker than all those of buthutin A, 9m, and 9n, which might be due to its too large spatial volume of aromatic ring in 9o.
In order to investigate the protective mechanism of these compounds against myocardial injury, the inhibition on NHE-1 activity was measured with the dpHi/min of H9c2 cardiomyocyte under the treatment of target compounds and positive control cariporide (Table 1). Among all tested compounds at 1 μM, compounds 7b, 9c, 9d, 9e, 9f, 9g, 9i, 9j, 9k, 9l, 9m, 9n, and 9o had the obvious inhibition effects on the NHE-1 activity. Furthermore, compounds 9k, 9m, and 9o exerted superior inhibitions on the NHE-1 activity than that of cariporide. As shown in Table 1, the ester-guanidine derivatives 13a–13e and five-carbon linker derivatives 9p–9u had no obvious NHE-1 inhibitory activities. Three-carbon linker derivatives 9a–9h showed weaker NHE-1 inhibitory activities than the corresponding four-carbon linker derivatives 9i–9o, for example, 9g versus 9n. Four-carbon linker derivatives with para hydroxyl substitution in benzene ring revealed the strongest inhibitory activities on NHE-1, such as buthutin A > 9i, 9j. Meanwhile, In amide-guanidine compounds with a p-hydroxyl group, when another substituent was introduced into its ortho or meta position, respectively, the meta substitution derivatives exerted a stronger NHE-1 inhibition than those with ortho substitution, such as 9k, 9m > 9l. All these results strongly supported the above SARs of myocardial injury protection.
To further evaluate the selectivity of buthutin A derivatives against other NHE isoforms, HEK293 cells expressing endogenous NHE-1, NHE-2 and NHE-3 [26, 27] were used to investigate the inhibitory effect on other NHEs activity. Reportedly, 3 µM cariporide was applied to inhibit most NHE-1 activity with minimal effect on other NHEs activities [28]. The results showed that the dpHi/min value were 1.33 ± 0.11 in model group and 0.44 ± 0.048 in 3 µM cariporide-treated group, which revealed that most NHE-1 activity was inhibited by 3 µM cariporide. However, additions of buthutin A, 9k, 9m and 9o to 3 µM cariporide could not further enhance the inhibition of 3 µM cariporide on the NHEs activities, with the dpHi/min values 0.39 ± 0.028, 0.38 ± 0.044, 0.42 ± 0.023 and 0.37 ± 0.014, respectively. Therefore, buthutin A, 9k, 9m and 9o could not obviously inhibit any other NHEs, mainly NHE-2 and NHE-3, which illustrated the specificity of inhibition on NHE-1 activity.
For clarifying the interaction mode of buthutin A derivatives in the active sites of NHE-1, molecular dockings were performed for compounds 9k, 9m, and 9o using Discovery Studio 2017 R2 software. Amazingly, compounds 9m and 9o exhibited much lower CDOCKER energies of –36.05 kcal/mol and –43.53 kcal/mol with NHE-1, respectively, in comparison to that (–34.71 kcal/mol) of buthutin A. Compounds 9m and 9o showed very close binding mode to buthutin A, both with three hydrogen bonds to Asp267 and Glu346 (Fig. 2). However, their additional hydroxyl group was stabilized by hydrogen bond interactions with ASP159 or HIS349 (Fig. 2), which should avail to increase the inhibitory activity. Further exploration of their dose–response relationships on NHE-1 was worth investigating.
3 Conclusions
In summary, twenty-nine buthutin derivatives (7a–7b, 9a–9u, and 13a–13e) were designed and synthesized, and their structures were well characterized by 1H-NMR, 13C-NMR, HR-ESI–MS analyses. These compounds were tested for their NHE-1 inhibitions and protective effects on cardiomyocyte injury for the first time. Among them, compounds 9d, 9f, 9k, 9m, and 9n displayed more potent protective effects against Dox-induced H9c2 cardiomyocyte injury than Dex and buthutin A, and compounds 9k, 9m, and 9o exerted notable inhibitions on the NHE-1 activity surpassing that of positive control cariporide. What is more, compounds 9k, 9m, 9o and buthutin A all exhibited the specificity on NHE-1 inhibition. Molecular modelling studies suggested the ability of compounds 9m and 9o to establish interactions with three hydrogen bonds to Asp267 and Glu346 of NHE-1, but also the ability with much lower CDOCKER energies than buthutin A and cariporide. Further, it could be concluded that all compounds had a similar structure–activity relationship between the inhibitory activities on NHE-1 and the protective effects against myocardial injury. The presences of amide group, four-carbon linker, and para hydroxyl benzene ring have been identified as pivotal pharmacophore for above two observed pharmacological actions, while both 3-methoxy-4-hydroxy-phenyl ring and 3,4-dihydroxy-phenyl ring also are conducive to the aforementioned activity. However, it could be inferred that the mechanism of myocardial protection was not entirely caused by NHE-1 inhibition, and that other mechanisms should also be involved, since some compounds (9a, 9b, 9u, 13a) had cardioprotective effects but no NHE-1 inhibitory activities. To sum up, based on the promising results observed in both cell-based assessments, these derivatives merit further investigation in the pursuit of developing cardioprotective agents.
4 Experimental section
4.1 General
A Waters Acquity UPLC Class I/Xevo G2 QTOF mass spectrometer (Milford, MA, USA) was used to obtain the high-resolution ESI–MS (HR-ESI–MS) data. 1H-NMR and 13C-NMR spectra were acquired on a Bruker Avance III 400 spectrometer spectrometer (Karlsruhe, Germany) with TMS as an internal standard. Melting points were determined using an X-4 digital micro-melting tester (Beijing Teck Instrument Co., Ltd.). Chromatographic analysis for purity determination (HPLC) was achieved on a Ultimate®XB-C18; 4.6 × 250 mm; 5 μm; column (Welch Materials, Inc. Shanghai, China). Gradient elution was performed starting at 5% MeOH increasing to 100% MeOH at 20 min, and detection was conducted at 254 nm. The ratio of peak areas in the chromatograms was used to express the purity in percentage.
4.2 Synthetic procedures
4.2.1 General procedure for the synthesis of intermediates 2
To a 100 mL flask, hydroxybenzoic acids 1 (0.1 mmol) and acetic anhydride (10 mL) were added. While the mixture was stirred, concentrated sulfuric acid (0.15 mL) was drip in. The reaction mixture was refluxed at 80 ℃ for 2 h. By adding 60 mL distilled water, the white precipitate was formed, and then was filtered and dried to obtain acetoxybenzoic acid intermediate 2.
4.2.2 General procedure for the synthesis of intermediates 4
Methyl isothioureide sulfate (3, 0.025 mmol), 30% K2CO3 solution (12.5 mL), and di-tert-butyl dicarbonate (0.1 mol) were dissolved in CH2Cl2 (25 mL) and stirred for 24 h at room temperature. The crude product was washed with water, and was purified by silica gel column chromatography using petroleum ether/ethyl acetate (4:1, v/v) as eluent to give compound 4 (Yield 98%) as an off- white solid.
4.2.3 General procedure for the synthesis of intermediates 5 and 10
A solution of 4 (7.5 mmo1) in CH2Cl2 (20 mL) was added in batches to a alkyl diamine (1, 3-propylene diamine, or 1, 4-butyric diamine, or 1, 5-pentylenediamine) (0.03 mol). The reaction mixture was stirred at 50 ℃ for 4 h. The crude product was washed with water to give series compounds 5. Series compounds 10 was prepared following the method described for the preparation of series compounds 5, employing 3-amino-propanol or 4-amino-1-butanol instead of alkyl diamine.
4.2.4 General procedure for the synthesis of intermediates 6a–6c, 8a–8u, and 11a–11e
Intermediates 2 (0.04 mol, 1.0 eq) and 5 (1.0 eq) were dissolved in DCM (20 mL) or DMF (20 mL) solution. To this solution, DCC (1.2 eq) and DMAP (0.1 eq) were added. The resulting reaction mixture was stirred for 24 h at room temperature. The reaction progress was detected by TLC. After completion of the reaction, a large amount of by-product DCU would be precipitated in the solution and filtered. Excess solvent of filtrate was removed by evaporation under reduced pressure to obtain the corresponding amide products 8a–8u. The method described above was used to synthesize amide products 11a–11e, employing series compounds 10 instead of series compounds 5. Similarly, amide products 6a–6c were also synthesized using series compounds 5 with 2-picolinic acid, nicotinic acid, isonicotinic acid, respectively.
4.2.5 Procedure for the synthesis of 7a–7c
A mixture of compounds 6a–6c (0.41 mmol, 1.0 eq) and zinc bromide (2.1 mmol, 5.0 eq) was stirred in DCM at room temperature for 10 h. The reaction progress was detected by TLC. After the completion of the reaction, the organic phase was washed with water, and further purified over Sephadex LH-20 eluted with MeOH to obtain target compounds 7a–7c.
N-(4-guanidinobutyl)-2-pyridinecarboxamide (7a). Yield 72%, white solid. Mp: 171–173 ℃; 1H NMR (D2O, 400 MHz) δ 8.55 (m, 1H, ArH), 7.95 (m, 2H, ArH), 7.56 (m, 1H, ArH), 3.41 (t, J = 6.4 Hz, 2H), 3.17 (t, J = 6.6 Hz, 2H), 1.62 (m, 4H); 13C NMR (100 MHz, D2O) δ 167.2, 156.6, 149.0, 148.7, 138.4, 127.1, 122.3, 40.6, 38.9, 25.6, 25.2; HR-ESI-MS (positive mode) m/z: 236.1515 [M + H]+ (calculated for C11H18N5O, 236.1511); Purity (HPLC): 95.1%.
N-(4-guanidinobutyl)-3-pyridinecarboxamide (7b). Yield 85%, white solid. Mp: 190–192 ℃; 1H NMR (D2O, 400 MHz) δ 8.76 (d, J = 1.7 Hz, 1H, ArH), 8.60 (dd, J = 1.7, 5.0 Hz, 1H, ArH), 8.10 (m, 1H, ArH), 7.51 (dd, J = 7.7, 5.0 Hz, 1H, ArH), 3.34 (t, J = 6.1 Hz, 2H), 3.14 (t, J = 6.4 Hz, 2H), 1.59 (m, 4H); 13C NMR (100 MHz, D2O) δ 168.0, 156.6, 150.8, 146.7, 136.3, 130.2, 124.3, 40.6, 39.3, 25.5, 25.2; HR-ESI-MS (positive mode) m/z: 236.1517 [M + H]+ (calculated for C11H18N5O, 236.1511); Purity (HPLC): 97.7%.
N-(4-guanidinobutyl)-4-pyridinecarboxamide (7c). Yield 71%, white solid. Mp: 108–110 ℃; 1H NMR (D2O, 400 MHz) δ 8.62 (d, J = 6.2 Hz, 2H, ArH), 7.64 (d, J = 6.2 Hz, 2H, ArH), 3.38 (t, J = 6.3 Hz, 2H), 3.17 (t, J = 6.5 Hz, 2H), 1.62 (m, 4H); 13C NMR (100 MHz, D2O) δ 168.6, 156.6, 149.4 (2C), 142.2, 121.5 (2C), 40.6, 39.3, 25.4, 25.2; HR-ESI-MS (positive mode) m/z: 236.1524 [M+H]+ (calculated for C11H18N5O, 236.1511); Purity (HPLC): 99.7%.
4.2.6 Procedure for the synthesis of 9a–9u
Compounds 8a–8u (1.16 mmol) and trifluoroacetic acid (1.16 mmol) were dissolved in Dichloromethane (10 mL). The resulting reaction mixture was heated at 50 ℃ for 24 h. After completion of the reaction, allowed it to room temperature and filtered. The crude product was recrystallized from MeOH to yield the final compounds 9a–9u.
N-(3-guanidinobutyl)-2-hydroxybenzamide (9a). Yield 75%, white solid. Mp: 118–120 ℃; 1H NMR (D2O, 400 MHz) δ 7.66 (d, J = 7.9 Hz, 1H, ArH), 7.43 (t, J = 7.6 Hz, 1H, ArH), 6.97 (m, 2H, ArH), 3.45 (t, J = 6.6 Hz, 2H), 3.24 (t, J = 6.6 Hz, 2H), 1.87 (qui, J = 6.6 Hz, 2H); 13C NMR (100 MHz, D2O) δ 170.8, 158.0, 157.6, 134.9, 129.1, 120.9, 117.9, 117.4, 39.6, 37.5, 28.5; HR-ESI-MS (positive mode) m/z: 237.1378 [M + H]+ (calculated for C11H17N4O2, 237.1352); Purity (HPLC): 98.6%.
N-(3-guanidinobutyl)-3-hydroxybenzamide (9b). Yield 82%, white solid. Mp: 130–132 ℃; 1H NMR (D2O, 400 MHz) δ 7.35 (t, J = 7.8 Hz, 1H, ArH), 7.25 (d, J = 7.8 Hz, 1H, ArH), 7.17 (s, 1H, ArH), 7.05 (d, J = 7.8 Hz, 1H, ArH), 3.43 (t, J = 6.7 Hz, 2H), 3.24 (t, J = 6.7 Hz, 2H), 1.87 (qui, J = 6.7 Hz, 2H); 13C NMR (100 MHz, D2O) δ 171.9, 158.0, 157.0, 136.5, 131.5, 120.24, 120.20, 115.1, 39.9, 38.2, 28.8; HR-ESI-MS (positive mode) m/z: 237.1378 [M + H]+ (calculated for C11H17N4O2, 237.1352); Purity (HPLC): 99.2%.
N-(3-guanidinobutyl)-4-hydroxybenzamide (9c). Yield 80%, white oil. 1H NMR (D2O, 400 MHz) δ 7.63 (d, J = 8.6 Hz, 2H, ArH), 6.90 (d, J = 8.6 Hz, 2H, ArH), 3.39 (t, J = 6.8 Hz, 2H), 3.21 (t, J = 6.8 Hz, 2H), 1.84 (m, 2H); 13C NMR (100 MHz, D2O) δ 170.4, 159.1, 156.7, 129.1 (2C), 125.3, 115.3 (2C), 38.6, 36.8, 27.5; HR-ESI-MS (positive mode) m/z: 237.1374 [M + H]+ (calculated for C11H17N4O2, 237.1352); Purity (HPLC): 97.8%.
N-(4-guanidinobutyl)-3-methoxy-4-hydroxybenzamide (9d). Yield 77%, white oil. 1H NMR (D2O, 400 MHz) δ 7.20 (m, 2H, ArH), 6.86 (d, J = 8.2 Hz, 1H, ArH), 3.81 (s, 3H), 3.36 (t, J = 6.7 Hz, 2H), 3.19 (t, J = 6.7 Hz, 2H), 1.82 (qui, J = 6.7 Hz, 2H); 13C NMR (100 MHz, D2O) δ 170.8, 158.0, 149.8, 148.3, 126.6, 122.1, 116.1, 111.9, 56.9, 40.1, 38.3, 29.1; HR-ESI-MS (positive mode) m/z: 267.1474 [M + H]+ (calculated for C12H19N4O3, 267.1457); Purity (HPLC): 99.4%.
N-(3-guanidinobutyl)-2,4-dihydroxybenzamide (9e). Yield 83%, white solid. Mp: 178–180 ℃; 1H NMR (D2O, 400 MHz) δ 7.58 (d, J = 8.7 Hz, 1H, ArH), 6.43 (dd, J = 2.4, 8.7 Hz, 1H, ArH), 6.37 (d, J = 2.4 Hz, ArH), 3.41 (t, J = 6.7 Hz, 2H) 3.22 (t, J = 6.6 Hz, 2H), 1.85 (m, 2H); 13C NMR (100 MHz, D2O) δ 169.6, 160.6, 160.0, 156.5, 129.3, 107.8, 107.6, 102.6, 38.6, 36.3, 27.6; HR-ESI-MS (positive mode) m/z: 253.1297 [M + H]+ (calculated for C11H17N4O3, 253.1301); Purity (HPLC): 99.8%.
N-(3-guanidinobutyl)-3,4-dihydroxybenzamide (9f). Yield 74%, white oil. 1H NMR (D2O, 400 MHz) δ 7.10 (br.s, 1H, ArH), 7.03 (d, J = 7.0 Hz, 1H, ArH), 6.76 (d, J = 7.0 Hz, 1H, ArH), 3.22 (br.s, 2H), 3.05 (br.s, 2H), 1.69 (br.s, 2H); 13C NMR (100 MHz, D2O) δ 169.4, 156.4, 147.7, 143.6, 125.2, 120.1, 115.3, 114.5, 38.6, 36.8, 27.6; HR-ESI-MS (positive mode) m/z: 253.1331 [M + H]+ (calculated for C11H17N4O3, 253.1301); Purity (HPLC): 97.8%.
N-(3-guanidinobutyl)-3,5-dihydroxybenzamide (9g). Yield 72%, white solid. Mp: 68–70 ℃; 1H NMR (D2O, 400 MHz) δ 6.68 (d, J = 2.2 Hz, 2H, ArH), 6.48 (t, J = 2.2 Hz, 1H, ArH), 3.35 (t, J = 6.8 Hz, 2H), 3.18 (t, J = 6.7 Hz, 2H), 1.81 (m, 2H); 13C NMR (100 MHz, D2O) δ 170.2, 157.0 (2C), 156.7, 136.2, 106.2 (2C), 105.8, 38.6, 36.9, 27.4; HR-ESI-MS (positive mode) m/z: 253.1348 [M + H]+ (calculated for C11H17N4O3, 253.1301); Purity (HPLC): 99.3%.
N-(3-Guanidinobutyl)-3,4,5-trihydroxybenzamide (9h). Yield 73%, white solid. Mp: 77–79 ℃; 1H NMR (D2O, 400 MHz) δ 6.74 (s, 2H, ArH), 3.23 (t, J = 6.8 Hz, 2H), 3.08 (t, J = 6.8 Hz, 2H), 1.71 (qui, J = 6.8 Hz, 2H); 13C NMR (100 MHz, D2O) δ 169.6, 156.6, 144.6 (2C), 136.2, 124.8, 107.3 (2C), 38.7, 36.9, 27.6; HR-ESI-MS (positive mode) m/z: 269.1272 [M + H]+ (calculated for C11H17N4O4, 269.1250); Purity (HPLC): 92.8%.
N-(4-guanidinobutyl)-2-hydroxybenzamide (9i). Yield 80%, white oil. 1H NMR (400 MHz, CD3OD) δ 7.60 (d, J = 7.4 Hz, 1H, ArH), 7.20 (t, J = 8.2 Hz, 1H, ArH), 6.73 (m, 2H, ArH), 3.27 (t, J = 6.2 Hz, 2H), 3.06 (t, J = 6.3 Hz, 2H), 1.51 (m, 4H); 13C NMR (100 MHz, CD3OD) δ 171.2, 161.3, 158.8, 134.8, 129.0, 120.2, 118.6, 117.2, 42.2, 39.9, 27.8, 27.4; HR-ESI-MS (positive mode) m/z: 251.1523 [M + H]+ (calculated for C12H19N4O2, 251.1508); Purity (HPLC): 96.1%.
N-(4-guanidinobutyl)-3-hydroxybenzamide (9j). Yield 83%, white solid. Mp: 84–86 ℃; 1H NMR (400 MHz, CD3OD) δ 7.17 (m, 3H, ArH), 6.87 (br.s, 1H, ArH), 3.32 (br.s, 2H), 3.15 (br.s, 2H), 1.58 (br.s, 4H); 13C NMR (100 MHz, CD3OD) δ 170.6, 158.9, 158.7, 137.1, 130.7, 119.6, 119.1, 115.3, 42.1, 40.2, 27.8, 27.3; HR-ESI-MS (positive mode) m/z: 251.1530 [M + H]+ (calculated for C12H19N4O2, 251.1508); Purity (HPLC): 96.0%.
N-(4-guanidinobutyl)-4-hydroxy-3-methoxybenzamide (9k). Yield 85%, white solid. Mp: 105–107 ℃; 1H NMR (400 MHz, D2O) δ 7.28 (d, J = 2.0 Hz, 1H, ArH), 7.24 (dd, J = 2.0, 8.2 Hz, 1H, ArH), 6.89 (d, J = 8.2 Hz, 1H, ArH), 3.83 (s, 3H), 3.32 (d, J = 6.2 Hz, 2H), 3.15 (d, J = 6.4 Hz, 2H), 1.59 (br.s, 4H); 13C NMR (100 MHz, D2O) δ 169.9, 156.6, 148.4, 147.1, 125.7, 120.8, 115.0, 110.9, 55.7, 40.6, 39.2, 25.6, 25.2; HR-ESI-MS (positive mode) m/z: 281.1634 [M + H]+ (calculated for C13H21N4O3, 281.1614); Purity (HPLC): 95.4%.
N-(4-guanidinobutyl)-2,4-dihydroxybenzamide (9l). Yield 76%, white solid. Mp: 74–76 ℃; 1H NMR (400 MHz, D2O) δ 7.45 (d, J = 8.8 Hz, 1H, ArH), 6.36 (dd, J = 2.4, 8.8 Hz, 1H, ArH), 6.27 (d, J = 2.4 Hz, 1H, ArH), 3.24 (br.s, 2H) 3.09 (d, J = 5.7 Hz, 2H), 1.53 (br.s, 4H); 13C NMR (100 MHz, D2O) δ 169.6, 160.7, 159.8, 156.6, 129.6, 108.5, 107.8, 102.8, 40.7, 38.7, 25.6, 25.2; HR-ESI-MS (positive mode) m/z: 267.1454 [M + H]+ (calculated for C12H19N4O3, 267.1457); Purity (HPLC): 95.1%.
N-(4-guanidinobutyl)-3,4-dihydroxybenzamide (9m). Yield 73%, white solid. Mp: 157–159 ℃; 1H NMR (400 MHz, CD3OD) δ 7.21 (d, J = 2.0 Hz, 1H, ArH), 7.12 (dd, J = 8.3, 2,0 Hz, 1H, ArH), 6.73 (d, J = 8.3 Hz, 1H, ArH), 3.24 (br.s, 2H) 3.10 (br.s, 2H), 1.51 (m, 4H); 13C NMR (100 MHz, CD3OD) δ 170.5, 158.7, 150.1, 146.2, 127.1, 120.8, 116.1, 115.9, 42.1, 40.2, 27.8, 27.2; HR-ESI-MS (positive mode) m/z: 267.1450 [M + H]+ (calculated for C12H19N4O3, 267.1457); Purity (HPLC): 95.2%.
N-(4-guanidinobutyl)-3,5-dihydroxybenzamide (9n). Yield 83%, white solid. Mp: 81–83 ℃; 1H NMR (400 MHz, D2O) δ 6.68 (d, J = 2.2 Hz, 2H, ArH), 6.49 (t, J = 2.2 Hz, 1H, ArH), 3.30 (t, J = 6.2 Hz, 2H), 3.14 (t, J = 6.3 Hz, 2H), 1.57 (br.s, 4H); 13C NMR (100 MHz, D2O) δ 170.1, 157.0 (2C), 156.6, 136.4, 106.1 (2C), 105.8, 40.6, 39.2, 25.5, 25.2; HR-ESI-MS (positive mode) m/z: 267.1451 [M + H]+ (calculated for C12H19N4O3, 267.1457); Purity (HPLC): 98.9%.
N-(4-Guanidinobutyl)-3,4,5-trihydroxybenzamide (9o). Yield 71%, white solid. Mp: 102–104 ℃; 1H NMR (400 MHz, D2O) δ 6.77 (s, 2H, ArH), 3.18 (br.s, 2H), 3.02 (br.s, 2H), 1.46 (br.s, 4H); 13C NMR (100 MHz, D2O) δ 169.5, 156.5, 144.6 (2C), 136.1, 124.9, 107.2 (2C), 40.6, 39.2, 25.6, 25.2; HR-ESI-MS (positive mode) m/z: 283.1425 [M + H]+ (calculated for C12H19N4O4, 283.1406); Purity (HPLC): 96.0%.
N-(5-guanidinopentyl)-2-hydroxybenzamide (9p). Yield 82%, white oil. 1H NMR (400 MHz, D2O) δ 7.79 (d, J = 7.4 Hz, 1H, ArH), 7.38 (t, 1H, J = 7.4 Hz, ArH), 7.11 (m, 2H, ArH), 3.51 (t, J = 6.5 Hz, 2H), 3.12 (t, J = 7.4 Hz, 2H), 1.81 (m, 4H), 1.57 (m, 2H); 13C NMR (100 MHz, D2O) δ 169.7, 156.9 (2C), 133.8, 128.1, 119.9, 116.9, 116.7, 39.2, 39.0, 27.8, 26.3, 22.9; HR-ESI-MS (positive mode) m/z: 265.1678 [M + H]+ (calculated for C13H21N4O2, 265.1665); Purity (HPLC): 97.6%.
N-(5-guanidinopentyl)-3-hydroxybenzamide (9q). Yield 86%, white oil. 1H NMR (400 MHz, CD3OD) δ 7.26 (m, 3H, ArH), 6.95 (d, J = 7.0 Hz, 1H, ArH), 3.39 (t, J = 7.0 Hz, 2H), 3.20 (t, J = 7.0 Hz, 2H), 1.66 (m, 4H), 1.46 (m, 2H); 13C NMR (100 MHz, CD3OD) δ 170.5, 158.9, 158.7, 137.3, 130.1, 119.5, 119.0, 115.3, 42.4, 40.6, 30.2, 29.6, 25.0; HR-ESI-MS (positive mode) m/z: 265.1668 [M + H]+ (calculated for C13H21N4O2, 265.1665); Purity (HPLC): 99.9%.
N-(5-guanidinopentyl)-4-hydroxybenzamide (9r). Yield 79%, white oil. 1H NMR (400 MHz, CD3OD) δ 7.72 (d, J = 8.8 Hz, 2H, ArH), 6.84 (t, J = 8.8 Hz, 2H, ArH), 3.39 (t, J = 7.0 Hz, 1H), 2.94 (t, J = 7.6 Hz, 2H), 1.69 (m, 4H), 1.46 (m, 2H); 13C NMR (100 MHz, CD3OD) δ 170.2, 162.1, 158.8, 130.2 (2C), 126.5, 116.1 (2C), 40.6, 40.4, 30.1, 28.2, 24.8; HR-ESI-MS (positive mode) m/z: 265.1672 [M + H]+ (calculated for C13H21N4O2, 265.1665); Purity (HPLC): 96.7%.
N-(5-guanidinopentyl)-4-hydroxy-3-methoxybenzamide (9s). Yield 85%, white oil. 1H NMR (400 MHz, CD3OD) δ 7.45 (d, J = 2.0 Hz, 1H, ArH), 7.37 (dd, J = 2.0, 8.2 Hz, 1H, ArH), 6.85 (d, J = 8.2 Hz, 1H, ArH), 3.91 (s, 3H), 3.39 (t, J = 7.0 Hz, 2H), 3.19 (t, J = 7.1 Hz, 2H), 1.65 (m, 4H), 1.45 (m, 2H); 13C NMR (100 MHz, CD3OD) δ 170.0, 158.7, 151.2, 148.8, 126.9, 122.0, 115.9, 111.9, 56.5, 42.4, 40.7, 30.3, 29.6, 25.1; HR-ESI-MS (positive mode) m/z: 295.1788 [M + H]+ (calculated for C14H23N4O3, 295.1770); Purity (HPLC): 95.4%.
N-(5-guanidinopentyl)-3,5-dihydroxybenzamide (9t). Yield 78%, white solid. Mp: 88–90 ℃; 1H NMR (400 MHz, CD3OD) δ 6.72 (d, J = 2.2 Hz, 2H, ArH), 6.44 (t, J = 2.2 Hz, 1H, ArH), 3.36 (t, J = 7.1 Hz, 2H), 3.19 (t, J = 7.1 Hz, 2H), 1.65 (m, 4H), 1.44 (m, 2H); 13C NMR (100 MHz, CD3OD) δ 170.7, 159.9(2C), 158.7, 138.0, 106.7(2C), 106.5, 42.4, 40.6, 30.1, 29.5, 25.0; HR-ESI-MS (positive mode) m/z: 281.1626 [M + H]+ (calculated for C13H21N4O3, 281.1614); Purity (HPLC): 99.8%.
N-(5-guanidinobutyl)-3,4,5-trihydroxybenzamide (9u). Yield 72%, white solid. Mp: 93–95 ℃; 1H NMR (400 MHz, D2O) δ 6.80 (s, 2H, ArH), 3.22 (t, J = 6.9 Hz, 2H), 3.05 (t, J = 6.9 Hz, 2H), 1.49 (m, 4H), 1.28 (m, 2H); 13C NMR (100 MHz, D2O) δ 169.7, 156.5, 144.7 (2C), 136.1, 125.2, 107.3 (2C), 40.8, 39.5, 27.8, 27.4, 23.0; HR-ESI-MS (positive mode) m/z: 297.1602 [M+H]+ (calculated for C13H21N4O4, 297.1563); Purity (HPLC): 96.6%.
4.2.7 General procedure for the synthesis of intermediates 12a–12e
To a solution of compounds 11a–11e in MeOH (10 mL), 12% NaOH (10 mL) solution was added. The resulting mixture was stirred at room temperature for 10 min. The reaction progress was detected by TLC. After concentration, the crude product was obtained and purified by silica gel column chromatography using ethyl acetate as eluent to give compounds 12a–12e.
4.2.8 Procedure for the synthesis of 13a–13e
Compounds 13a–13e were synthesized by using the same way for 7a–7c. Nevertheless, compounds 12a–2e was used instead of compounds 6a–6c.
3-Guanidinopropyl 3-hydroxybenzoate (13a). Yield 77%, white solid. Mp: 140–142 ℃; 1H NMR (400 MHz, CD3OD) δ 7.52 (dt, J = 7.9, 1.4 Hz, 1H, ArH), 7.45 (dd, J = 2.4, 1.4 Hz, 1H, ArH), 7.31 (t, J = 7.9 Hz, 1H, ArH), 7.05 (ddd, J = 7.9, 2.4, 1.4 Hz, 1H, ArH), 4.40 (t, J = 6.2 Hz, 2H), 3.39 (t, J = 6.9 Hz, 2H), 2.08 (m, 2H); 13C NMR (100 MHz, CD3OD) δ 168.0, 158.9, 158.8, 132.5, 130.7, 121.6, 121.4, 117.1, 63.1, 39.5, 29.2; HR-ESI-MS (positive mode) m/z: 238.1196 [M + H]+ (calculated for C11H16N3O3, 238.1192); Purity (HPLC): 99.8%.
3-Guanidinopropyl 4-hydroxybenzoate (13b). Yield 73%, white oil. 1H NMR (400 MHz, CD3OD) δ 7.90 (d, J = 8.7 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 4.36 (t, J = 5.9 Hz, 2H), 3.40 (t, J = 6.8 Hz, 2H), 2.08 (m, 2H); 13C NMR (100 MHz, CD3OD) δ 168.0, 163.0, 158.1, 132.6 (2C), 121.7, 116.0 (2C), 62.7, 39.4, 28.8; HR-ESI-MS (positive mode) m/z: 238.1189 [M + H]+ (calculated for C11H15N3O3, 238.1192); Purity (HPLC): 99.8%.
3-Guanidinopropyl 4-hydroxy-3-methoxybenzoate (13c). Yield 88%, white oil. 1H NMR (400 MHz, CD3OD) δ 7.58 (dd, J = 8.3, 1.9 Hz, 1H, ArH), 7.53 (d, J = 1.9 Hz, 1H, ArH), 6.86 (d, J = 8.3 Hz, 1H, ArH), 4.37 (t, J = 6.1 Hz, 2H), 3.91 (s, 3H), 3.40 (t, J = 6.9 Hz, 2H), 2.09 (m, 2H); 13C NMR (100 MHz, CD3OD) δ 168.1, 158.1, 154.4, 148.4, 125.0 (2C), 115.8, 113.1, 62.8, 56.5, 39.4, 28.9; HR-ESI-MS (positive mode) m/z: 268.1302 [M + H]+ (calculated for C12H18N3O4, 268.1297); Purity (HPLC): 99.8%.
3-Guanidinopropyl 3,5-dihydroxybenzoate (13d). Yield 79%, white solid. Mp: 70–72 ℃; 1H NMR (400 MHz, CD3OD) δ 6.95 (d, J = 2.2 Hz, 2H, ArH), 6.50 (t, J = 2.2 Hz, 1H, ArH), 4.37 (t, J = 6.2 Hz, 2H), 3.38 (t, J = 6.9 Hz, 2H), 2.07 (m, 2H); 13C NMR (100 MHz, CD3OD) δ 168.0, 159.7 (2C), 158.5, 132.9, 108.8 (2C), 108.3, 63.0, 39.4, 29.1; HR-ESI-MS (positive mode) m/z: 254.1147 [M + H]+ (calculated for C11H16N3O4, 254.1141); Purity (HPLC): 98.3%.
4-Guanidinobutyl 4-hydroxybenzoate (13e). Yield 78%, white solid. Mp: 79–81 ℃; 1H NMR (400 MHz, CD3OD) δ 7.88 (d, J = 8.8 Hz, 2H, ArH), 6.85 (d, J = 8.8 Hz, 2H, ArH), 4.31 (t, J = 6.1 Hz, 2H), 3.29 (t, J = 7.0 Hz, 2H), 1.85 (m, 2H), 1.76 (m, 2H); 13C NMR (100 MHz, CD3OD) δ 168.2, 163.1, 158.1, 132.6 (2C), 121.9, 116.0 (2C), 65.1, 42.0, 26.8, 26.3; HR-ESI-MS (positive mode) m/z: 252.1362 [M + H]+ (calculated for C12H18N3O3, 252.1348); Purity (HPLC): 99.7%.
4.3 Molecular docking
Molecular docking was carried out using Discovery Studio 2017 R2 (Accelrys, San Diego, USA). Docking studies were conducted with DS CDOCKER program. The cryoelectron microscopy (cryo-EM) structure of NHE-1 from Homo sapiens (PBD ID: 7DSX) was obtained from the Protein Data Bank. All water molecules and allied ligand were removed followed by protein preparation protocol with the CHARMm force field. The simulated annealing parameters were set as follows: heating steps and cooling steps were set to 2000 and 5000, respectively, while heating and cooling temperatures were set to 700 and 300, respectively. Other parameters were kept as default. Ten top-ranked conformations for each docked compound were retained and visually inspected for binding pattern analysis using Discovery Studio Visualizer.
4.4 Biological activity
4.4.1 Cell culture and cell viability assay
The H9c2 cardiomyocyte (rat myocardial cell line) and HEK 293 cell (Human Embryonic Kidney 293 cells) were obtained from ATCC (American type culture collection) and grown in Dulbecco’s modifed Eagle’s medium (DMEM) (Wuhan Pricella Biotechnology Co., Ltd.) with 10% fetal bovine serum (FBS, Wuhan Pricella Biotechnology Co., Ltd.) and 1% penicillin–streptomycin (Beijing Solarbio Science & Technology Co., Ltd.) at 37 ℃ in a humidified incubator consisting of 5% CO2 and 95% air. After reaching about 70–80% confluence, the H9c2 cardiomyocyte was digested by trypsinization (0.25% Trypsin–EDTA solution, Beijing Solarbio Science&Technology Co., Ltd) every three days. After seeded into the 96-well culture plates for 24 h with 3000 cells/well, the cells were divided into control group, Dox treated group, Dex treated group, buthutin A treated group, and compounds-treated group. The cells in Dox-treated group were treated with 1 μM Dox for 24 h, while the Dex-treated group, buthutin A treated group, and compounds-treated groups were treated with 20 μM Dex and 1 μM Dox, 1 μM buthutin A and 1 μM Dox, and 1 μM target compound and 1 μM Dox for 24 h, respectively. At the same time, control group cells were incubated continuously with the normal medium. After treatment, the MTT solution (Beijing Solarbio Science&Technology Co., Ltd.) with the final concentration of 0.5 mg/ml was added into each well and incubated for 4 h at 37 ℃. After formazan product in each well was dissolved, the absorbance of each well was measured at 570 nm by using a microplate reader (BioTek Instruments, Inc.) [21]. The protection ratio of each compound was calculated as (Acompound–Ablank)/(Acontrol–Ablank) × 100%.
4.4.2 Measurement of intracellular pH (pHi)
The pHi of cells was measured by the pH-sensitive fluorescent probe [2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein, acetoxymethyl ester (BCECF-AM, Beyotime)] [29]. After seeded into the 96-well plate for 24 h with 10,000 cells/well, H9c2 cardiomyocyte was incubated with Krebs Solution for 30 min, and then the diluted BCECF-AM with the final concentration of 1 μM was added into each well for another 30 min. The BCECF-AM was washed up with Krebs Solution and incubated for 30 min, the fluorescence value was measured with Spectra Max iD3 (Molecular Devices) at 480 nm and 440 nm for excitation and 530 nm for emission, then fluorescence intensity ratio (FIR) was calculated as FIR480 nm/FIR440 nm × 100%. The pHi standard curve was made as follows: five wells with almost the same values of FIR were selected, and added the high-potassium solution containing 4 mg/L nigericin sodium salt (Beijing psaitong Biotechnology Co., Ltd) with different pH gradients (6.5, 6.8, 7.1, 7.4 and 7.7), and incubated for 8 min; then the pHi standard curve was derived based on the pH value and corresponding FIR value. Then, the H9c2 cardiomyocyte was divided into control group, model group, cariporide treated group (1 μM), buthutin A treated group (1 μM), and compounds treated group (1 μM). Except the control group, the cells of other groups were incubated with 25 mM NH4Cl diluted with Krebs Solution for 3 min, and then the cells were washed with sodium-free Krebs Solution to clean NH4Cl. Target compound diluted with sodium-free Krebs Solution was added to the cells and cultured for 10 min. Then the target compound was washed with sodium-containing Krebs Solution, and the FIR value of each well was detected. The pHi values of each group were calculated according to the standard pHi curve. The activity of NHE-1 is expressed as the rate of pHi change per minute (dpHi/min) from the sodium-free to sodium-containing phase. In addition, the HEK293 cells were divided into control group, model group, cariporide (3 µM)-treated group, cariporide (3 µM) and buthutin A (1 µM)-treated group, cariporide (3 µM) and 9k (1 µM)-treated group, cariporide (3 µM) and 9m (1 µM)-treated group, and cariporide (3 µM) and 9o (1 µM)-treated group. All these groups received the intracellular pH (pHi) measurement to estimate the NHEs activity.
4.5 Statistical analysis
The data of this research were expressed as mean ± SEM, which were analyzed with SPSS 19.0 and GraphPad Prism 7.0. All tests were carried out at least in quintuplicate. The statistical comparisons in the different group were performed using one-way ANOVA followed by Tukey's post hoc test. Differences were considered to have statistical significance at p < 0.05.
Notes
Acknowledgements
The authors are thankful to the Analysis and Testing Center at Tianjin University of Technology for providing analytical facilities.
Author contributions
Yuan Liu, Xin-Hao Hua and Fa-Qi Wang: Carrying out experiments, writing original draft. Shu-Han Yang: data curation. Li-Ning Wang: Methodology, software. Yun-Sheng Xu and Chen-Yue Shao: Biological screening, review/editing. Xiang-Bo Gou: Writing the article, bioactivity evaluation. Yu-Ming Liu: Conceptualization, supervision, designing experiments, review/editing. The authors read and approved the final manuscript.
Funding
This work was financially supported by the National Natural Science Foundation of China (NSFC) Youth Project (No. 82204397).
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its Supporting Information files.
Declarations
Competing interests
The authors declare no competing interests.
References
-
1.Medina AJ, Pinilla OA, Portiansky EL, Caldiz CI, Ennis IL, et al. Silencing of the Na+/H+ exchanger 1 (NHE-1) prevents cardiac structural and functional remodeling induced by angiotensin II. Exp Mol Pathol 2019;107: 1-9. CrossRef PubMed Google Scholar
-
2.Pan G, Cui B, Han M, Lin L, Li Y, Wang L, et al. Puerarin inhibits NHE1 activity by interfering with the p38 pathway and attenuates mitochondrial damage induced by myocardial calcium overload in heart failure rats: Puerarin inhibits NHE1 activity. Acta Biochim Biophys Sin 2024;56(2): 270-9. CrossRef PubMed Google Scholar
-
3.Yeves AM, Ennis IL. Na+/H+ exchanger and cardiac hypertrophy. Hipertens Riesgo Vasc 2020;37(1): 22-32. CrossRef PubMed Google Scholar
-
4.Chen S, Overberg K, Ghouse Z, Hollmann MW, Weber NC, Coronel R, et al. Empagliflozin mitigates cardiac hypertrophy through cardiac RSK/NHE-1 inhibition. Biomed Pharmacother 2024;174: 116477. CrossRef PubMed Google Scholar
-
5.Jaballah M, Mohamed IA, Alemrayat B, Al-Sulaiti F, Mlih M, Mraiche F. Na+/H+ exchanger isoform 1 induced cardiomyocyte hypertrophy involves activation of p90 ribosomal s6 kinase. PLoS ONE 2015;10(4): e0122230. CrossRef PubMed Google Scholar
-
6.Mohamed IA, Gadeau AP, Fliegel L, Lopaschuk G, Mlih M, Abdulrahman N, et al. Na+/H+ exchanger isoform 1-induced osteopontin expression facilitates cardiomyocyte hypertrophy. PLoS ONE 2015;10(4): e0123318. CrossRef PubMed Google Scholar
-
7.Carvalho C, Santos RX, Cardoso S, Correia S, Oliveira PJ, Santos MS, Moreira PI. Doxorubicin: the good, the bad and the ugly effect. Curr Med Chem 2009;16: 3267-85. CrossRef PubMed Google Scholar
-
8.Carvalho FS, Burgeiro A, Garcia R, Moreno AJ, Carvalho RA, Oliveira PJ. Doxorubicin-induced cardiotoxicity: From bioenergetic failure and cell death to cardiomyopathy. Med Res Rev 2014;34: 106-35. CrossRef PubMed Google Scholar
-
9.Rawat PS, Jaiswal A, Khurana A, Bhatti JS, Navik U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed Pharmacother 2021;139: 111708. CrossRef PubMed Google Scholar
-
10.Wenningmann N, Knapp M, Ande A, Vaidya TR, Ait-Oudhia S. Insights into doxorubicin-induced cardiotoxicity: molecular mechanisms, preventive strategies, and early monitoring. Mol Pharmacol 2019;96: 219-32. CrossRef PubMed Google Scholar
-
11.Zhang P-P, Lu H, Wu Y, Lu D-B, Li C-G, Yang X-D, et al. COX5A alleviates doxorubicin-induced cardiotoxicity by suppressing oxidative stress, mitochondrial dysfunction and cardiomyocyte apoptosis. Int J Mol Sci 2023;24: 10400. CrossRef PubMed Google Scholar
-
12.Sangweni NF, Moremane M, Riedel S, van Vuuren D, Huisamen B, Mabasa L, et al. The prophylactic effect of pinocembrin against doxorubicin-induced cardiotoxicity in an in vitro H9c2 cell model. Front Pharmacol 2020;11: 1172. CrossRef PubMed Google Scholar
-
13.Yin Y, Niu Q, Hou H, Que H, Mi S, Yang J, et al. PAE ameliorates doxorubicin-induced cardiotoxicity via suppressing NHE1 phosphorylation and stimulating PI3K/AKT phosphorylation. Int Immunopharmacol 2022;113: 109274. CrossRef PubMed Google Scholar
-
14.Tebbi CK, London WB, Friedman D, Villaluna D, De Alarcon PA, Constine LS, et al. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin’s disease. J Clin Oncol 2007;25: 493-500. CrossRef PubMed Google Scholar
-
15.Lee BH, Seo HW, Yi KY, Lee S, Lee S, Yoo SE. Effects of KR-32570, a new Na+/H+ exchanger inhibitor, on functional and metabolic impairments produced by global ischemia and reperfusion in the perfused rat heart. Eur J Pharmacol 2005;511: 175-82. CrossRef PubMed Google Scholar
-
16.Avkiran M, Cook AR, Cuello F. Targeting Na+/H+ exchanger regulation for cardiac protection: a RSKy approach. Curr Opin Pharmacol 2008;8: 133-40. CrossRef PubMed Google Scholar
-
17.Cuadrado I, Oramas-Royo S, González-Cofrade L, Amesty Á, Hortelano S, Estévez-Braun A. Labdane conjugates protect cardiomyocytes from doxorubicin-induced cardiotoxicity. Drug Dev Res 2023;84: 84-95. CrossRef PubMed Google Scholar
-
18.Alzahrani AM, Rajendran P, Veeraraghavan VP, Hanieh H. Cardiac protective effect of kirenol against doxorubicin-induced cardiac hypertrophy in H9c2 cells through Nrf2 signaling via PI3K/AKT pathways. Int J Mol Sci 2021;22(6): 3269. CrossRef PubMed Google Scholar
-
19.Gao H, Yang X, Gu X, Zhu YZ. Synthesis and biological evaluation of the codrug of leonurine and aspirin as cardioprotective agents. Bio Med Chem Lett 2016;26: 4650-4. CrossRef PubMed Google Scholar
-
20.Zhu YZ, Wu W, Zhu Q, Liu X. Discovery of Leonuri and therapeutical applications: from bench to bedside. Pharmacol Therapeut 2018;188: 26-35. CrossRef PubMed Google Scholar
-
21.Li C, Gou X, Gao H. Doxorubicin nanomedicine based on ginsenoside Rg1 with alleviated cardiotoxicity and enhanced antitumor activity. Nanomedicine 2021;16(29): 2587-604. CrossRef PubMed Google Scholar
-
22.Wan M, Yin K, Yuan J, Ma S, Xu Q, Li D, et al. YQFM alleviated cardiac hypertrophy by apoptosis inhibition and autophagy regulation via PI3K/AKT/mTOR pathway. J Ethnopharmacol 2022;285: 114835. CrossRef PubMed Google Scholar
-
23.Liu YM, Fan JJ, Wang LN. Discovery of guanidine derivatives from Buthus martensii Karsch with metal-binding and cholinesterase inhibition properties. Molecules 2021;26: 6737. CrossRef PubMed Google Scholar
-
24.Salamouni NSE, Buckley BJ, Lee R, Ranson M, Kelso MJ, Yu H. Ion transport and inhibitor binding by human NHE1: Insights from molecular dynamics simulations and free energy calculations. J Phys Chem B 2024;128: 440-50. CrossRef PubMed Google Scholar
-
25.Dong Y, Gao Y, Ilie A, Kim D, Boucher A, Li B, et al. Structure and mechanism of the human NHE1-CHP1 complex. Nat Commun 2021;12: 3474. CrossRef PubMed Google Scholar
-
26.Roosterman D. Agonist-dependent and -independent dopamine-1-like receptor signalling differentially regulates downstream effectors. FEBS J 2014;281: 4792-804. CrossRef PubMed Google Scholar
-
27.Beloto-Silva O, Machado UF, Oliveira-Souza M. Glucose-induced regulation of NHEs activity and SGLTs expression involves the PKA signaling pathway. J Membrane Biol 2011;239: 157-65. CrossRef PubMed Google Scholar
-
28.Nolte AP, Chodisetti G, Yuan Z, Busch F, Riederer B, Luo M, et al. Na+ /H+ exchanger NHE1 and NHE2 have opposite effects on migration velocity in rat gastric surface cells. J Cell Physiol 2017;232(7): 1669-80. CrossRef PubMed Google Scholar
-
29.Cardoso VG, Gonçalves GL, Costa-Pessoa JM, Thieme K, Lins BB, Casare FAM, et al. Angiotensin II-induced podocyte apoptosis is mediated by endoplasmic reticulum stress/PKC-δ/p38 MAPK pathway activation and trough increased Na+/H+ exchanger isoform 1 activity. BMC Nephrol 2018;19: 179. CrossRef PubMed Google Scholar
Copyright information
© The Author(s) 2025
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
